MAPPATURA FISICA E GENETICA
Strategie di mappatura
Acquisire la completa conoscenza della organizzazione, struttura
e funzione del genoma umano e' certamente una delle piu' ambiziose
mete scientifiche.
Esaminando l'organizzazione delle sequenze di DNA nel genoma umano,
si e' stimato che circa l'1-5% delle 3x109 pb che costituiscono
il patrimonio genetico aploide codifica per proteine.
Una meta primaria per incominciare a comprendere l'organizzazione
del genoma e' di ottenere una serie di diagrammi di mappa descrittivi
di ciascun cromosoma umano a una risoluzione sempre piu' fine.
Per costruire una mappa genomica e' necessario avere a disposizione
frammenti di DNA, precedentemente isolati e inseriti in vettori
di clonaggio. I cloni ricombinanti sono successivamente ordinati
nelle loro rispettive localizzazioni sul cromosoma. Dopo il completamento
della mappatura, il passaggio successivo e' quello di determinare
la sequenza di basi di ciascun frammento ordinato. Questi stessi
scopi sono stati fissati da un programma di ricerca, chiamato
Progetto Genoma Umano
- il completamento di una mappa genetica ad alta risoluzione (1cM)
del genoma umano;
- completamento di una serie di mappe fisiche ad alta risoluzione
di tutti i cromosomi umani;
- acquisizione di una collezione di cloni di DNA ordinati che
coprano l'intero genoma;
- determinazione della sequenza nucleotidica completa di un genoma
di riferimento;
- mappatura di tutti i geni.
Una mappa genomica descrive l'ordine di locus genetici o di marcatori
e la distanza compresa tra loro su ciascun cromosoma.
Le mappe possono essere divise in genetiche e fisiche a seconda
dei metodi usati per costruirle e della unita' di misura adoperate
per valutare la distanza tra due marcatori.
Mappe genetiche per linkage
Le mappe genetiche per linkage sono mappe costruite mediante
associazione genetica, determinando la frequenza con cui due marcatori
sintenici (ossia associati e quindi localizzati sullo stesso cromosoma)
sono ereditati insieme. Nelle mappe genetiche non e' necessario
conoscere la localizzazione sul cromosoma dei markers studiati,
in quanto nella costruzione di queste mappe si cerca di svelare
l'associazione tra due o piu' locus. Locus che sono molto vicini
sul cromosoma hanno una probabilita' maggiore di essere ereditati
insieme rispetto a loci distanti.
Studi genetici su famiglie per determinare quanto frequentemente
due o piu' coppie di alleli sono ereditati insieme, permettono
la costruzione di mappe genetiche in cui la distanza tra due geni
e' misurata in cM (in onore al genetista americano Thomas Hunt
Morgan). Un cM, o unita' di mappa genetica, e' definito come la
distanza tra due geni per i quali un prodotto della meiosi su
cento e' ricombinante.
Ponendo in un grafico la distanza tra due loci, misurata in cM,
in funzione della frequenza di ricombinazione, si osserva una
deviazione dalla proporzionalita' diretta, per distanze tra due
loci maggiori di 40-50 cM (FIG.1). Due fattori possono essere
causa della relazione non lineare tra frequenza di ricombinazione
e distanza di mappa genetica: i crossing-over multipli che possono
avvenire tra due loci e l'interferenza. L'interferenza positiva
indica l'effetto per cui un crossing-over puo' ridurre la probabilita'
di un secondo crossing-over nelle sue vicinanze. Questa interferenza
sembra essere limitata a crossing-over che avvengono sullo stesso
braccio di un cromosoma.
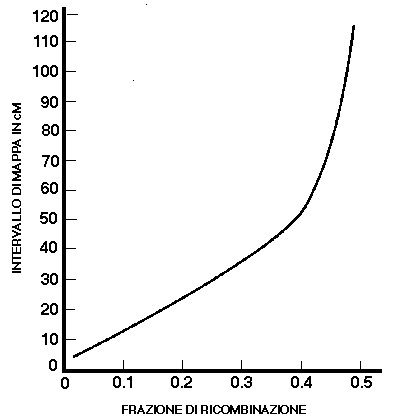
Fig. 1 Relazione tra distanza di mappa genetica e frequenza
di ricombinazione
Markers
Affinche' un marker possa essere usato per costruire mappe
genetiche, esso deve essere polimorfico.
Qualsiasi caratteristica fisica o molecolare ereditata nella progenie
e che differisce tra gli individui di una popolazione e che abbia
una frequenza superiore all'1%, e' un potenziale marker genetico.
I markers possono essere o regioni di DNA esprimibili o segmenti
di DNA anonimi dei quali non si conosce niente, tranne che sono
polimorfici. I numerosi alleli per molecole MHC di classe I e
II, gli antigeni del gruppo sanguigno ABO (un individuo puo' essere
di gruppo sanguigno A, B, AB, o 0), la presenza di varianti enzimatiche
(es. forma A e B di G-6Pi-DH, distinguibili su gel di poliacrilamide
per diverso peso molecolare e mobilita' elettroforetica), sono
esempi di polimorfismo per sequenze di DNA codificante, il cui
pattern ereditario puo' essere facilmente seguito. Altri markers
invece sono in grado di definire un polimorfismo neutrale, ossia
variazioni all'interno di DNA non associate a effetti fenotipici
tra gli individui di una popolazione, poiche' spesso le mutazioni
cadono all'interno di introni o sono mutazioni silenti. Esempi
di quest'ultima categoria di markers sono i VNTR basati sugli
RFLPs. I VNTR (Variable Number of Tandem Repeats) sono sequenze
nucleotidiche note ripetute in tandem un numero variabile di volte,
precedute e seguite da sequenze di DNA a singola copia. Digerendo
il DNA genomico di diversi individui con enzimi di restrizione
che tagliano all'interno delle sequenze uniche che fiancheggiano
un locus VNTR, si evidenzieranno dopo corsa elettroforetica del
prodotto di digestione, frammenti di lunghezza diversi a seconda
del numero di ripetizioni. Polimorfismi di lunghezza degli enzimi
di restrizione, che sono alla base degli RFLPs (Restriction Fragment
Lenght Polymorphisms), possono essere dovuti anche alla presenza/assenza
nel genoma di un individuo di siti di taglio per enzimi di restrizione.
Mutazioni puntiformi all'interno della sequenza riconosciuta da
un enzima di restrizione hanno spesso come unico effetto la mancata
digestione del DNA da parte di quell'enzima (evidenziabile dalla
diversa dimensione dei frammenti dopo corsa elettroforetica).
In ogni caso queste variazioni tra gli individui possono essere
usate come marcatori per costruire mappe genetiche di linkage.
Nel 1993/94 il Genethon, usando come markers dei microsatelliti,
ossia sequenze da 1 a 4 pb ripetute in tandem un numero variabile
di volte e intersperse nel genoma, ha costruito una nuova mappa
genetica. Sono stati mappati 2066 dinucleotidi (AC)n, ottenendo
1266 intervalli per una distanza di 3690 cM. La distanza media
tra i markers e' di 2.9cM, ma il 56% e' ad una distanza di 1cM.
Queste mappe possono essere usate per mappare in particolare geni
responsabili di malattie monogeniche. Recenti mappe consensus
di alcuni cromosomi hanno una distanza tra i markers di circa
7-10cM.
Inizialmente i markers sono stati isolati in maniera casuale da
libraries genomiche e successivamente mappati su un dato cromosoma.
Oggi nuove tecniche permettono di isolare markers DNA cromosoma-specifici
partendo da libraries cromosoma specifiche. Queste possono essere
realizzate mediante l'uso di uno strumento, chiamato FACS (Fluorence
Actived Cell Sorter), che permette di separare i vari cromosomi
di una sospensione, colorata con fluorocromi, in base alla intensita'
di fluorescenza che a sua volta e' direttamente proporzionale
alla grandezza del cromosoma. Il cromosoma isolato e' digerito
con enzimi di restrizione e i frammenti sono clonati in specifici
vettori. Sempre per ottenere markers per una regione subcromosomica
specifica (ad es. una banda cromosomica) si puo' usare la tecnica
della microdissezione. Un micromanipolatore dotato di aghi molto
sottili e' usato per tagliare una particolare banda da un preparato
metafasico colorato con tecniche di colorazione differenziale
e osservato al microscopio. La regione di interesse e' cosi' isolata
e clonata mediante PCR. Il DNA cosi' ottenuto puo' essere usato
come sonda per l'ibridazione e per identificare grandi inserti
di DNA presenti nelle libraries genomiche.
Mappe fisiche
Differenti tipi di mappa fisica, che descrivono l'ordine e
la distanza tra due marcatori misurata in numero di nucleotidi
(bp), variano nel loro grado di risoluzione, ossia nella precisione
della localizzazione di una sonda. I metodi usati nella mappatura
fisica si possono dividere in due categorie:
- metodi a bassa risoluzione;
- metodi ad alta risoluzione.
Mappa fisica a bassa risoluzione
I sistemi di mappatura piu' usati utilizzano gli ibridi cellulari
e la tecnica della ibridazione in situ.
Mappatura mediante ibridi cellulari
Cellule somatiche ibride interspecifiche derivano dalla fusione
di cellule provenienti da specie differenti.
Gli ibridi piu' usati per la mappatura derivano dalla fusione
di cellule umane e di roditore, comunemente topo o hamster, indotta
da sostanze chimiche, come il polietilenglicole, in grado di creare
dei pori a livello della membrana plasmatica.
Le cellule ibride che inizialmente tendono a perdere in maniera
casuale e progressiva cromosomi umani, in seguito si stabilizzano
fino a contenere il set cromosomico del roditore e il cromosoma
umano che assicura un vantaggio selettivo all'ibrido, insieme
a cromosomi ritenuti casualmente.
Dopo aver selezionato le cellule ibride e averle caratterizzate
dal punto di vista cariotipico, si allestisce un pannello di ibridi,
dato dall'insieme delle cellule ibride il cui contenuto cromosomico
umano nel complesso corrisponde all'intero set cromosomico dell'uomo.
Tale pannello e' usato per studi di mappatura.
Gli ibridi cellulari sono stati utilizzati inizialmente per mappare
geni di cui si conosceva il prodotto (proteine enzimatiche o strutturali).
Sugli estratti proteici delle cellule ibride si esegue una corsa
elettroforetica su gel. Il gel si usa in seguito o per far avvenire
una reazione enzimatica catalizzata dall'enzima il cui gene si
vuole mappare, in presenza di un cromogeno, o per far avvenire
(in caso di proteine non enzimatiche) una reazione di precipitazione
usando anticorpi contro la proteina stessa. Se la cellula ibrida
non ha ritenuto il cromosoma su cui mappa il gene codificante,
non si avra' reazione colorimetrica o di precipitazione.
Un limite di questa applicazione e' dato dal fatto che, essendo
il topo o l'hamster e l'uomo due specie affini, potrebbero esserci
problemi nel discriminare tra la reazione, colorimetrica o di
precipitazione, dovuta a proteine umane e quella relativa a proteine
murine.
Oggi, grazie all'avvento delle tecniche di DNA ricombinante, e'
possibile mappare qualsiasi segmento clonato indipendentemente
dalla sua espressione. Digerendo il DNA estratto dai cloni ibridi
con enzimi di restrizione, eseguendo un blotting e ibridando con
la sonda a disposizione, e' possibile localizzarla su un dato
cromosoma, calcolando l'indice di concordanza, ossia il rapporto
percentuale tra gli ibridi positivi per la sonda in esame e gli
ibridi totali. Usando un enzima in grado di discriminare tra la
banda di ibridazione umana e murina, si puo' riconoscere l'eventuale
segnale di ibridazione proveniente dalle cellule di roditore.
Un limite della mappatura mediante ibridi che contengono solo
cromosomi umani interi e' che non si puo' definire la posizione
subcromosomica. Questo limite puo' essere superato utilizzando
ibridi che contengono frammenti di un dato cromosoma, ottenuti
dalla fusione di cellule di roditore e cellule ibride contenenti
un solo cromosoma umano e irradiate, o ibridi ottenuti da cellule
parentali umane provenienti da pazienti con riarrangiamenti cromosomici.
Ibridazione in situ fluorescente (FISH)
L' ibridazione in situ non radioattiva permette di localizzare
direttamente una sonda, marcata in modo non radioattivo, sulla
corrispondente regione cromosomica.
Le sonde, clonate in plasmidi, fagi, cosmidi o YACs, possono essere
facilmente associate con particolari bande (identificabili attraverso
colorazioni citogenetiche), ottenendo una mappa detta citogenetica
o cromosomica. Il numero di paia di basi contenute in una banda
puo' essere solo stimato.
Fin' ora anche la migliore mappa cromosomica puo' essere usata
solo per localizzare frammenti di DNA in una regione di circa
10Mb, la misura di una tipica banda vista su un cromosoma. Tecniche
ad alta risoluzione, le quali permettono di ottenere cromosomi
in prometafase, possono incrementare la risoluzione di mappa di
circa 0.1Mb.
Se la sonda usata per l'ibridazione in situ e' un cDNA si ottiene
una mappa, detta mappa cDNA, che mostra la posizione di regioni
di DNA espresse (esoni) relative a particolari regioni o bande
cromosomiche. Queste regioni di DNA sono trascrivibili in RNAm.
Il cDNA e' sintetizzato in laboratorio usando RNAm come template;
questo cDNA puo' essere quindi mappato su regioni genomiche. Poiche'
esse rappresentano regioni genomiche espresse, i cDNA identificano
le parti del genoma con maggiore interesse biologico e medico.
Una mappa cDNA puo' fornire la localizzazione cromosomica per
geni le cui funzioni sono attualmente sconosciute. Integrando
mappe cDNA e mappe genetiche di linkage si puo' testare la localizzazione
di un ipotetico gene responsabile di una malattia.
Mappa fisica ad alta risoluzione
Con determinate tecniche si possono mappare sequenze di DNA
con una risoluzione da 1 pb a molte Mb. Le mappe fisiche ad alta
risoluzione sono fondamentalmente basate su due approcci:
- collezione di frammenti clonati di DNA provenienti da un intero
cromosoma o parti di esso (genoteche totali o parziali). Le mappe
possono essere prodotte identificando cloni che hanno inserti
di DNA che si sovrappongono. Si cerca di ottenere un contiguo,
ossia una serie di cloni overlappanti che coprano una intera regione
cromosomica. Questi cloni rappresentano la base di partenza per
analisi piu' dettagliate fino a livello di sequenza.
- analisi per elettroforesi di frammenti di DNA ottenuti mediante
digestione con enzimi di restrizione "rare cutter" che
producono macroframmenti. Per frazionare grossi frammenti di restrizione
prodotti e' necessario utilizzare una particolare tecnica di elettroforesi
in campi pulsati e diversamente orientati (PFGE da Pulsed field
gel elctrophoresis). Il risultato di questa tecnica e' dato dall'ordine
e distanza tra siti tagliati da suddetti enzimi. Questo approccio
produce mappe con maggiore continuita' e pochi gaps tra i frammenti
rispetto alle mappe costruite usando contigui, anche se la risoluzione
e' piu' bassa.
Esistono diversi vettori per il clonaggio molecolare come i plasmidi,
in grado di contenere frammenti fino a 10 Kb, fagi, che possono
contenere frammenti fino a 40 Kb, e cosmidi in grado di contenere
frammenti fino a 100 Kb.
Per la mappatura fisica su larga scala e' necessario organizzare
contigui che coprano estese regioni cromosomiche. Tale obiettivo
risulta tanto piu' facilmente realizzabile quanto piu' grandi
saranno i frammenti clonati.
Lo sviluppo dei cromosomi artificiali di lievito (YACs) come vettori
di clonaggio, mediante i quali e' possibile isolare frammenti
di DNA dell'ordine di grandezza di 1 Mb, ha dato un notevole contributo
alla soluzione di questo problema.
Per identificare YACs di una data regione cromosomica in modo
da costruire un contiguo, si possono usare delle sequenze nucleotidiche
note, lunghe da 200 a 500 pb, distribuite su tutta una regione
genomica o su tutto il genoma dette STS (Sequence Tagged Site).
E' stata costruita una mappa mostrando l'ordine e la distanza
degli STS. La mappa con gli STS puo' essere rappresentata elettronicamente
e conservata in una banca dati facilmente accessibile. Un STS
identifica in maniera univoca un qualsiasi tratto di DNA e pertanto
un laboratorio che voglia clonare uno specifico frammento genomico,
dovra' semplicemente consultare un computer per conoscere le due
sequenze STS che delimitano questo frammento. Usando due primers
corrispondenti alle due sequenze STS, si potra' amplificare mediante
PCR il frammento e successivamente clonarlo. Poiche' nella reazione
di amplificazione l'enzima usato non riesce ad amplificare frammenti
superiori a 1000 pb, i primers scelti, corrispondenti alle due
sequenze STS che delimitano il frammento da clonare, non devono
distare piu' di 1000 pb.
Poiche' ogni elemento mappato (cloni, contiguo, sequenza) e' definito
da un unico STS, la mappe con gli STS sono molto utili per identificare
e combinare dati di mappatura ottenuti da differenti strategie.
Per identificare tra le sequenze di DNA umano quelle corrispondenti
a geni sono usati particolari STS, chiamati ESTs (Expressed Sequence
Tags), ottenuti scrinando libraries di cDNA, che riconoscono sequenze
trascritte. Infatti una applicazione degli ESTs include la localizzazione
di geni lungo i cromosomi e l'identificazione di regioni codificanti
nel genoma umano.
Relazione tra mappa genetica e mappa fisica
La distanza della mappa genetica per le 3000Mb del genoma
umano e' di circa 3000cM e pertanto 1cM corrisponde approssimativamente
a una distanza di mappa fisica di 0.8Mb.
In realta' il rapporto delle distanze di mappa genetica e fisica
sui segmenti cromosomici spesso deviano da questo valore medio
a causa della localizzazione non casuale dei chiasmi. I segmenti
cromosomici contenenti gli "hot spots" di ricombinazione
mostrano una piu' alta frequenza di ricombinazione e per tanto
markers localizzati in questa zona sembrano piu' distanti del
reale. In generale c'e' una frequenza di crossing-over piu' alta
in corrispondenza delle regioni subtelomeriche rispetto a quelle
centromeriche.
YACs
Gli YACs (cromosomi artificiali di lievito) sono vettori di
clonaggio molto utili per la costruzione di mappe fisiche ad alta
risoluzione. Gli YACs permettono infatti di clonare larghe regioni
di DNA (da 100Kb a piu' di 1500Kb), risultando uno strumento utilissimo
per ottenere contigui di una data regione cromosomica. Piu' che
di vettori veri e propri gli YACs sono dei cromosomi artificiali
inseriti in una cellula ospite di lievito. Per potersi replicare
un cromosoma ha bisogno di strutture fondamentali:
una o piu' origini di replicazione per permettere la duplicazione
del DNA che contiene e strutture terminali, dette telomeri, che
assicurano la completa replicazione del DNA fino all'estremita'
di un cromosoma lineare. Cromosomi privi di telomeri si accorciano
leggermente ma costantemente ad ogni ciclo di replicazione cellulare
e tendono a rompersi alle estremita' per saldarsi ad altri cromosomi.
Analizzando questi requisiti si e' pensato di poter costruire
un cromosoma condensato, ossia una molecola lineare contenente
una origine di replicazione, un centromero (necessario per la
corretta segregazione dei cromosomi) e due telomeri agli estremi.
Tale minicromosoma si puo' replicare e restare come molecola lineare
all'interno del nucleo di una cellula eucariotica. Gli YACs sono
inseriti in cellule di lievito poiche' contengono le sequenze
ARS (sequenze che si replicano autonomamente) di lievito. Tali
cromosomi possono ospitare al loro interno frammenti di DNA grandi
fino a 1Mb. Per le grandi dimensioni degli inserti clonati, un
numero ridotto di cloni sara' necessario per coprire l'intero
genoma.
Nel sistema di mappatura, gli YACs ottenuti estraendo il DNA dalle
cellule di lievito che li contengono non sono usati, a differenza
di altri vettori come plasmidi e cosmidi, come sonde tal quali,
poiche' l'alto rapporto tra DNA di lievito e frammento di DNA
clonato renderebbe necessario l'utilizzo di una elevata quantita'
di DNA per aumentare l'efficienza del sistema di mappatura.
Per superare questo limite gli YACs sono usati come sonde per
esperimenti di FISH dopo essere stati amplificati con primer Alu,
sequenze di 300pb distribuite una volta circa ogni 5Kb nel genoma
umano, le quali permettono l'amplificazione specifica di sequenze
inter-Alu umane dal DNA di lievito. L'amplificazione aumenta l'efficienza
del segnale in esperimenti di FISH rispetto all'utilizzo della
stessa sonda non amplificata. L'efficienza della generazione di
sonde mediante Alu-PCR dipende dal numero delle sequenze Alu adeguatamente
spaziate che fiancheggiano le sequenze sito specifiche in ciascuno
YACs. Cloni YACs derivati da parti del genoma con un contenuto
particolarmente basso in sequenze Alu daranno un segnale meno
efficiente rispetto a cloni derivati da sequenze genomiche ricche
di Alu.
Allo stesso scopo si puo' utilizzare una tecnica di separazione
elettroforetica di lunghi frammenti di DNA, detta PFGE (pulse
field gel electrophoresis), che permette di separare i cromosomi
artificiali di lievito da quelli naturali. Nella PFGE larghi frammenti
di DNA ricavati dalla digestione di regioni genomiche possono
essere separati su gel d'agarosio attraversato da un campo elettrico
discontinuo. Il principio su cui si basa questa tecnica e' quello
di far ruotare le molecole di DNA all'interno del gel in modo
che anche molecole molto lunghe possano passare tra le maglie
della matrice semisolida e migrare piu' o meno velocemente secondo
la loro lunghezza. Si possono cosi' separare frammenti di DNA
lunghi da 50Kb a qualche Mb.
Gli YACs sono molto usati in citogenetica come sonde specifiche
di determinate regioni cromosomiche. Sono anche usati per isolare
e caratterizzare regioni coinvolte in malattie genetiche o per
caratterizzare punti di rottura sui cromosomi associate ad es.
a trasformazioni neoplastiche. Per questo scopo e' necessario
conoscere la posizione fisica degli YACs e verificare che non
siano chimerici.
Gli YACs chimerici sono YACs che contengono frammenti di DNA che
ibridizzano su differenti regioni del genoma. Questo puo' essere
dovuto o alla presenza di due frammenti diversi all'interno dello
stesso clone o alla presenza di elementi ripetuti all'interno
del genoma stesso. Per superare questi problemi si sta cercando
di utilizzare tecniche sempre piu' raffinate come quella che permette
di separare su gel di agarosio frammenti di DNA molto piccoli
da grandi inserti in maniera da ridurre la possibilita' di clonare
insieme nello stesso YAC questi due tipi di frammenti. Inoltre
si sta cercando di creare una banca dati, completa anche di immagini,
relativa a YACs dimostratisi non chimerici e omogeneamente distribuiti
lungo il genoma. Per ottenere YACs di una data regione cromosomica,
al fine di costruire un contiguo, si possono utilizzare dati sulla
distribuzione degli STS lungo il genoma. Utilizzando sempre gli
STS si possono ordinare gli YACs del contiguo. Due YACs che si
sovrappongono mostreranno un comune STS, mentre YACs che contengono
due o piu' STS potranno essere usati per estendere il contiguo.
Painting libraries cromosomiche e subcromosomiche
L'insieme di tutti i vettori ricombinanti, che nel complesso
coprono un intero cromosoma, rappresenta una libreria cromosoma
specifica.
I vettori di clonaggio piu' usati per la costruzione di una libreria
sono i cosmidi e gli YACs, che a differenza dei plasmidi e dei
fagi, consentono di inserire larghi frammenti di DNA.
Le libraries cromosomiche possono essere costruite o mediante
cell sorter (precedentemente descritto) o partendo da ibridi cellulari
uomo-hamster contenenti un solo cromosoma umano. Sottoponendo
il DNA dell'ibrido ad amplificazione genica usando come primers
sequenze Alu, specifiche dell'uomo, si amplifica la sola componente
umana dell'ibrido. IL prodotto dell'amplificazione, usato come
sonda per esperimenti di FISH su metafasi umane, colorera' solo
l'intero cromosoma umano contenuto nell'ibrido.
A causa dei riarrangiamenti che avvengono in vitro durante la
formazione degli ibridi, questi ultimi possono ritenere, in aggiunta
a cromosomi interi, uno o piu' frammenti cromosomici. Il contenuto
cromosomico umano dell'ibrido puo' essere caratterizzato ibridando
prodotti marcati di Alu-PCR dell'ibrido su metafasi umane. Gli
ibridi con frammenti cromosomici noti possono essere usati come
painting libraries subcromosomiche.
L'abbinamento tra painting libraries cromosomiche e subcromosomiche
con YACs specifici di una data regione cromosomica, permette di
caratterizzare in maniera fine riarrangiamenti cromosomici incontrati
in citogenetica clinica o tumorale e consente di seguire riarrangiamenti
avvenuti durante l'evoluzione. L'utilita' di questo abbinamento
e' particolarmente evidente quando la regione cromosomica riarrangiata
e' talmente piccola da non poter essere identificata tramite i
metodi convenzionali di bandeggio. Inoltre l'uso delle sole painting
libraries cromosomiche fornirebbe indicazioni solo sul cromosoma
riarrangiato e non sulla regione subcromosomica interessata e
sui punti di rottura, mentre l'analisi con i soli YACs di un dato
cromosoma richiederebbe molto tempo.
L'uso combinato delle painting libraries subcromosomiche e degli
YACs permette di restringere rispettivamente l'analisi ad una
regione definita e di definire in maniera precisa la regione cromosomica
riarrangiata. Questo puo' essere realizzato mediante esperimenti
di coibridazioni in cui le due sonde (quella relativa alla painting
librarie e quella relativa allo YAC), marcate per nick translation
in maniera diversa (una con biotina e l'altra con digossigenina),
sono ibridate insieme. Esse potranno essere distinte, in esperimenti
di FISH, sullo stesso preparato metafasico.