L'osservazione
che Gregorio Mendel fece alla metà del XIX secolo, nel
corso dei suoi primi esperimenti sui piselli, sarebbe diventata
in seguito un assioma per i genetisti. Mendel si era accorto che,
incrociando piante di pisello "pure" con semi lisci
e piante di pisello "pure" con semi grinzosi, si ottenevano
piante "ibride" equivalenti, ma tutte con semi lisci.
Il risultato era il medesimo sia che la pianta con piselli lisci
rappresentasse la generazione parentale maschile sia, inversamente,
che rappresentasse la generazione parentale femminile. Dunque,
Mendel aveva scoperto il Principio dell'equivalenza negli incroci
reciproci: il gene, indipendentemente dal genitore che lo trasmette
alla prole, si comporta sempre allo stesso modo. Nella storia
e nella pratica genetica l'importanza dell'osservazione di Mendel
non può certo essere sottovalutata. Essa è, infatti,
risultata valida per un gran numero di caratteri genetici, non
soltanto i piselli, ma anche i moscerini della frutta, i topi,
gli esseri umani e tutta una schiera di altri organismi. In retrospettiva,
comunque, pare che Mendel selezionò con cura i gruppi dei
caratteri dei piselli che segregavano chiaramente, mentre ci sono
parecchi altri caratteri negli stessi piselli, come in molte altre
specie, che non mostrano una ereditarietà mendeliana.
Una delle sfide
più importanti della genetica contemporanea è quella
di dimostrare quei caratteri e quelle condizioni che non seguono
l'ereditarietà secondo Mendel. E' in questa prospettiva
che il concetto di imprinting genomico ha assunto una crescente
importanza in quanto potrebbe fornire una spiegazione per una
serie notevolmente vasta di osservazioni sulle condizioni per
cui la trasmissione e l'espressione genetica non si conformano
alle previsioni dell'ereditarietà a gene singolo. Il termine
"imprinting" è stato usato per la prima volta
in biologia da Lorenz alla fine degli anni '30 per descrivere
alcune osservazioni sul comportamento animale: ci sono momenti
critici durante le prime fasi della vita in cui il comportamento
può essere modificato da particolari esperienze ed esposizioni,
come osservato quando degli anatroccoli subito dopo la schiusa
venivano indotti a riconoscere come madre un cane, qualora questo
rappresentasse la prima cosa che vedevano.
Il termine
"imprinting" è stato adoperato in generale per
indicare qualsiasi tipo di modificazione del comportamento conseguente
ad esperienze particolari. Più recentemente, si adopera
il termine di " imprinting genomico" per riferirsi all'espressione
differenziata di materiale genetico, a seconda che esso derivi
dal genitore di sesso maschile oppure dal genitore di sesso femminile.
L'imprinting genomico deve implicare modificazioni del DNA nucleare
di cellule somatiche affinché possa produrre delle differenze
fenotipiche; è un concetto questo, che contraddice l'assioma
mendeliano secondo il quale l'origine dell'informazione genetica
non influenza l'espressione dei geni. In questo contesto il termine
imprinting è usato anche per indicare che un qualcosa accade
durante il periodo "critico o sensibile" dello sviluppo.
Nel caso dell' imprinting genomico lo stadio in cui si formano
le cellule germinali, potrebbe rappresentare il periodo critico
durante il quale le informazioni genetiche vengono "etichettate"
o "marcate", vale a dire che esse vengono temporaneamente
modificate per permettere una espressione differenziata.
L'
imprinting genomico è un processo che, in maniera temporanea
e reversibile, lascia un' impronta di diverso tipo nei geni trasmessi
dal genitore di sesso maschile e in quelli trasmessi dal genitore
di sesso femminile. Di conseguenza, la prole che riceve geni marcati
dalla madre sarà geneticamente diversa da quella che riceve
geni marcati dal padre. In poche parole, è in qualche caso
importante il fatto che un gene venga trasmesso da un genitore
piuttosto che dall' altro. In realtà, sono note da molti
anni alcune eccezioni al principio della equivalenza degli ibridi
reciproci, ma in generale esse sono sempre state fatte rientrare
nell' una o nell' altra di due categorie.
Della
prima categoria fanno parte i caratteri legati ai geni localizzati
sui cromosomi sessuali, X e Y. Le femmine dei mammiferi hanno
due cromosomi sessuali X in tutti i nuclei cellulari, mentre nei
maschi questi cromosomi sono diversi e sono noti come X e Y. Tra
i numerosi caratteri determinati da geni presenti sul cromosoma
X possiamo ricordare il daltonismo e l'emofilia. La loro trasmissione
ereditaria segue uno schema ben definito, che non deve essere
necessariamente equivalente negli ibridi reciproci. Per esempio,
se un uomo daltonico ha figli da una donna non portatrice di daltonismo,
nessuno dei figli maschi sarà daltonico. Se la madre è
daltonica e il padre no, i figli maschi saranno invece tutti daltonici.
In entrambi i casi, le figlie saranno portatrici
del gene per il daltonismo, ma non saranno daltoniche. La trasmissione
ereditaria e la manifestazione del carattere legato al sesso dipende
dal sesso della prole, ma non direttamente dal sesso del genitore
che presenta quel carattere.
La
seconda categoria di incroci reciproci non equivalenti contempla
caratteri controllati da geni situati al di fuori del nucleo cellulare.
Alcuni oganelli subcellulari ( i mitocondri nelle cellule animali,
e i mitocondri e i cloroplasti nelle cellule vegetali ) possiedono
una propria informazione genetica; poiché vengono trasmessi
di generazione in generazione col citoplasma della cellula uovo
i loro geni vengono ereditati esclusivamente per via materna.
L' encefalomiopatia mitocondriale (una forma di malattia
neuromuscolare) nell'uomo mostra questo tipo di ereditarietà.
L'
imprinting genomico, il terzo tipo di eccezione al principio dell'
equivalenza degli incroci reciproci (e quello scoperto più
di recente), differisce nettamente dagli esempi riportati in precedenza.
I caratteri che esso influenza non sono necessariamente
determinati da geni localizzati sui cromosomi sessuali (anche
se possono esserlo) e neppure sono associati a organelli ereditati
per via materna. Le eccezioni legate al sesso e agli
Organelli materni si basano su un ineguale contributo genetico
da parte di ciascun genitore. Per contro, a causa dell'
imprinting genomico, anche se i genitori trasmettono alla prole
geni assolutamente identici, gli effetti di questi geni non saranno
uguali se sarà stato esercitato su di essi un imprinting
diverso.
A
suggerire l' esistenza di un imprinting genomico esistono cinque
tipi di osservazioni dedotte da studi effettuati sui topi e anche
sugli uomini:
1 ) osservazioni su risultati di esperimenti di tipo trasferimento
pronucleare nei topi,
2 ) fenotipi dei triploidi umani;
3 ) espressione di certe disomie cromosomiche nel topo e nell'uomo;
4 ) espressione fenotipica delle deficenze cromosomiche nei topi
e nell'uomo (in relazione a sindrome specifiche e a tumori maligni);
5 ) espressione di materiale genetico transgenico in topi transgenici.
1 ) osservazioni su risultati di esperimenti di trasferimento
pronucleare nei topi.
In
buona parte, l' interesse attuale attorno ai caratteri influenzati
da un imprinting genomico deriva dagli esperimenti di D. Solter,
M. Azim H. Surani et al.. Questi autori hanno
messo a punto una elegante tecnica " chirurgica " estremamente
fine, il trasferimento di nuclei, che permette di scambiare
fisicamente l ' informazione genetica di un embrione di topo
con quello di un altro embrione. Questa tecnica è
resa possibile dal fatto che, dopo la fecondazione dell' uovo
da parte dello spermatozoo, ma prima che le cellule dell' embrione
comincino a dividersi, il nucleo della cellula uovo e quello
dello spermatozoo rimangono separati per breve tempo nel citoplasma
dello zigote (il che suggerisce già ruoli diversi). Ciascuno
dei due nuclei è visibile al microscopio ottico e ha una
dimensione e una posizione caratteristiche.
Per
mezzo di una sottilissima pipetta di vetro, sono riusciti ad asportare
selettivamente dallo zigote o il nucleo appartenente all' uovo
o quello appartenente allo spermatozoo (o entrambi ). Sono poi
riusciti a sostituire il nucleo rimosso con un nucleo proveniente
da un altro zigote e hanno trovato che, sostituendo un nucleo
derivato da uno spermatozoo con uno avente la stessa provenienza
o facendo la stessa operazione con un nucleo ottenuto da una cellula
uovo, gli embrioni risultanti si sviluppavano in modo completo
ed erano indistinguibili dai topi normali dello stesso ceppo.
Mediante
trasferimento di nuclei, è stato possibile dare origine
ad embrioni con entrambi le serie di cromosomi di un unico genitore.
Volendo sapere in che modo questi embrioni si sarebbero sviluppati,
hanno prodotto alcuni "ginogenoti " (embrioni con due
serie complete di cromosomi materni) e "androgenoti "(embrioni
con due serie complete di cromosomi paterni) e ne hanno confrontato
lo sviluppo con quello di embrioni normali. E' importante
notare che, essendo stati incrociati fra loro da molte generazioni,
i topi utilizzati in questi esperimenti avevano, sia nei maschi,
sia nelle femmine, serie identiche di cromosomi (tranne, ovviamente,
il cromosoma Y, mancante nelle femmine, e il cromosoma X, presente
in unica copia nei maschi). Se lo sviluppo di un embrione
di topo dipende esclusivamente dal corredo genico che l' animale
possiede, non dovrebbe in teoria importare che un topo riceva
tutti i geni da un solo genitore: ginogenoti, androgenoti e topi
normali dovrebbero tutti svilupparsi in modo identico. In realtà,
invece, questi embrioni non si sviluppano nello stesso modo.
Lo
sviluppo sia degli individui con due serie di geni di origine
materna, sia di quelli con due serie di geni di origine paterna
non giunge a termine: generalmente si arresta dopo che la
divisione ha prodotto qualche decina di cellule. Quando
riesce a proseguire, come capita di tanto in tanto, i ginogenoti
e gli androgenoti mostrano diverse e interessanti anomalie. Nei
ginogenoti che hanno raggiunto lo stadio più avanzato,
gli embrioni presentano aberrazioni relativamente poco importanti,
ma la placenta e il sacco vitellino (strutture essenziali per
il nutrimento dell' embrione) sono assai scarsamente sviluppati.
Negli androgenoti che hanno raggiunto lo stadio di
sviluppo più avanzato accade esattamente il contrario:
il sacco vitellino e la placenta sembrano più vicini
alla normalità, mentre gli embrioni sono gracili
e poco sviluppati. Entrambe le situazioni sono letali. Questo
tipo di lavoro di trapianto pronucleare suggerisce che sia i cromosomi
derivati dalla madre sia quelli derivati dal padre sono necessari
per uno sviluppo embrionale normale: entrambi forniscono un indispensabile
contributo all' embrione e alla placenta.
Poiché
la sequenza del DNA nei cromosomi dei ginogenoti, degli androgenoti
e degli embrioni normali sono sempre uguali, e' stata formulata
l' ipotesi che i geni fossero stati in qualche modo modificati
o marcati in maniera diversa in conseguenza della loro origine
materna o paterna. Tra i geni di origine paterna, alcuni, importanti
per lo sviluppo embrionale, sembravano inattivi, mentre tra i
geni di origine materna parevano inattivi quelli aventi una importanza
critica per la formazione del sacco vitellino e della placenta.
Nell'uomo
alcune patologie possono essere considerate omologhe ai risultati
degli esperimenti di trasferimento pronucleare nei topi: la "
mola idatiforme "(malformazione placentare) ed il "teratoma"
(tumore di origine embrionale). La mola completa viene
riscontrata nelle gravidanze prive di embrione in cui la placenta
e' sviluppata in maniera abnorme e disorganizzata; essa ha due
sets aploidi di cromosomi derivanti dal padre, e pertanto puo'
essere definita androgenetica. Di solito si è riscontrato
un raddoppiamento del corredo cromosomico di uno spermatozoo 23-X
normale, ma a volte le moli sono il risultato di dispermia. Al
contrario, i teratomi sono tumori embrionali dei tessuti provenienti
da tutti e tre i foglietti germinali, ma privi di tessuto placentale.
E' stato dimostrato che i teratomi ovarici hanno due sets aploidi
di cromosomi derivati dalla madre, il che sta ad indicare che
sono ginogenetici.
2 ) Fenotipi dei triploidi umani
I
triploidi derivano dal raddoppio del normale contributo di un
genitore e mostrano un funzionamento diverso del materiale genetico
paterno e materno. Il triploide con due corredi paterni ed uno
materno (diandria) viene tipicamente osservato sottoforma di una
placenta cistica, con parziali cambiamenti molari. Se è
presente il feto, di solito questo ha potuto sopravvivere grazie
al mosaicismo ed ha un aspetto caratteristico: testa abbastanza
grande, corpo piccolo e affusolato, forte ritardo della crescita
intrauterina e sindattilia.
Il triploide con due corredi materni
ed uno paterno (ginoide) ha una placenta poco sviluppata, priva
di cambiamenti cistici, e' presente un marcato sottosviluppo correlato
probabilmente al fallimento della placenta. Queste
osservazioni supportano la tesi secondo la quale le informazioni
genetiche paterne giocano un ruolo particolarmente critico circa
lo sviluppo ed il mantenimento delle membrane e della placenta.
3 ) espressione di alcune disomie cromosomiche nel topo
e nell'uomo.
Un'
altra brillante dimostrazione dell' imprinting è stata
data da B. M. Cattanach il quale da anni si interessava al fenomeno
della non equivalenza di certi incroci reciproci. Per studiare
questi caratteri, Cattanach si servì di ceppi di topi che
presentavano una o più coppie di cromosomi fusi: topi,
cioè, in cui alcuni cromosomi sono fisicamente saldati
e, pertanto, non hanno la possibilità di separarsi nel
corso della meiosi,. Di conseguenza una cellula che si divide
potrà ricevere occasionalmente due copie di uno stesso
cromosoma, mentre un' altra cellula non ne riceverà alcuna.
Se
un uovo con due copie di un cromosoma viene fecondato da uno spermatozoo
che non possiede quel cromosoma, l' embrione che ne risulterà
conterrà il numero normale di cromosomi, ma entrambe le
copie di un cromosoma proverranno dalla madre. Analogamente entrambe
le copie saranno di origine paterna se lo spermatozoo conterrà
i due cromosomi e la cellula uovo non ne conterrà alcuno.
Secondo il principio dell' equivalenza negli ibridi reciproci,
tutti gli animali che derivano da tali incroci dovrebbero essere
identici. Ma Cattanach ha inequivocabilmente dimostrato, con i
suoi esperimenti, che tale principio non vale per diversi cromosomi
di topo.
Per
esempio, egli ha trovato che i topi che avevano ricevuto entrambe
le copie del cromosoma 11 da un unico genitore erano radicalmente
diversi da quelli normali, per la taglia e per il peso. Allevati
nella medesima condizione, i figli con due cromosomi 11 di origine
materna avevano dimensioni anormalmente piccole, mentre quelli
che avevano ricevuto quei due cromosomi dal padre avevano dimensioni
aumentate.
Una
situazione che riguarda l' uomo, simile alle disomie uniparentali
del topo, è rappresentata da due casi di fibrosi cistica.
In entrambi questi casi, i bambini affetti hanno ricevuto tutte
e due i cromosomi 7 dalla madre; i padri non sembrano portatori
di fibrosi cistica e la loro paternità è stata dimostrata
utilizzando markers del DNA. La disomia uniparentale ha
un certo numero di implicazioni degne di nota; in questo contesto
va sottolineato che entrambi i bambini (un maschio ed una femmina)
presentavano un forte ritardo di crescita intrauterina e di crescita
post-natale, il che ricorda le osservazioni fatte circa la disomia
nel topo.
Gli
esperimenti di Cattanach sono valsi a dimostrare un altro punto
importante: gli effetti dell' imprinting genomico non persistono
nella generazione successiva, il che significa che il fenomeno
non provoca una modificazione permanente del cromosoma. Un topo
di piccole dimensioni e di sesso maschile, con entrambe le copie
del cromosoma 11 provenienti dalla madre, dà vita, di norma,
a prole di dimensioni normali. In un modo o
nell' altro, dunque, i geni ricevuti dalla madre devono essere
privati dall' impronta femminile e devono venire nuovamente contrassegnati
come geni maschili.
Non
è chiaro se i maggiori effetti fenotipici delle disomie
uniparentali sono causate dalla duplicazione (cioè dalla
presenza di due cromosomi dello stesso genitore) o dalla deficienza
(cioè dalla mancanza di un cromosoma di un genitore.
4) espressione fenotipica delle deficenze cromosomiche nell'uomo
(in relazione a sindrome specifiche e a tumori maligni)
La
Sindrome di Prader-Willi, caratterizzata da ipotonia nell' infanzia,
obesità dovuta ad iperfagia, ipogonadismo, mani e piedi
piccoli ed una facies peculiare, è dovuta ad una delezione
del cromosoma 15q11-13. E' stato determinato che il
cromosoma 15 deleto è derivato dal padre in quasi tutti,
se non tutti, i casi osservati. E' stato dimostrato che
in parecchi casi di Prader-Willi in cui non è possibile
dimostrare una delezione, entrambi i cromosomi 15 sono stati ereditati
dalla madre. Questi dati suggeriscono che è la mancanza
del cromosoma 15 paterno (o almeno della parte critica della regione
15q11-13 ) a causare il fenotipo della Prader-Willi.
Una
situazione analoga e' stata riscontrata in pazienti affetti da
Sindrome di Angelman. La Sindrome di Angelman è caratterizzata
da movimenti ripetitivi, simmetrici, atassici e da una disposizione
all' allegria, al riso frequente, con guance rosse e bocca larga
( i bambini affetti vengono descritti come " bambini pupazzo
"). La metà circa dei pazienti ha delezioni
evidenziabili citogeneticamente del cromosoma 15q11-13, simili
a quelle osservate nella sindrome di Prader-Willi. Tuttavia, nella
sindrome di Angelman, le delezioni coinvolgono il cromosoma 15
ereditato dalla madre.
E'
sorprendente che due malattie con quadri clinici così diversi
possano essere collegate ad un imprinting differenziale di regioni
genomiche localizzate sullo stesso cromosoma. Ma, contrariamente
ai topi anormalmente grossi o piccoli, che Cattanach ha ottenuto
attraverso i suoi esperimenti, la sindrome di Prader-Willi e quella
di Angelman non si possono spiegare facilmente come facce opposte
di una stessa medaglia, causate da un eccesso o da una carenza
degli stessi prodotti genici. Infatti le due sindromi non sono
dovute a mutazioni dello stesso locus, ma a mutazioni di geni
diversi fisicamente associati che subiscono un imprintig diverso.
Lo studio di questi mutanti ha permesso di individuare una regione
definita centro di imprinting sul cromosoma 15 (vedi oltre)
L'insorgenza di alcune forme di cancro infantile, tra
cui il rabdomiosarcoma dell'embrione (un tumore che colpisce i
muscoli), il tumore di Wilms (una forma di carcinoma renale) e
l'osteosarcoma (una forma di cancro delle ossa) e' legata all'imrpinting
delle regioni genomiche in cui sono localizzati gli oncogenei
corrispondenti.
Per
spiegare come l' imprinting genomico possa trovarsi coinvolto
in queste malattie, e' opportuno illustrare brevemente come si
pensa che compaiono le suddette forme di tumore. L'insorgenza
del tumore e' la conseguenza di un accumulo di successive mutazioni
in geni specifici all' interno di una singola cellula. Per esempio,
sul cromosoma 11 si trova un gene, Rd, che fa parte di una famiglia
di geni conosciuti come oncogeni recessivi, o geni che sopprimono
i tumori o anti-oncogeni. Senza il prodotto di Rd una cellula
muscolare si trasforma in una cellula neoplastica che poi evolve
in un rabdomiosarcoma. Dato che ogni nucleo cellulare
contiene due copie del cromosoma 11, prima che la cellula
si trasformi devono essere disattivate entrambe le copie di Rd.
La prima copia può essere disattivata in molteplici modi,
il più semplice dei quali è una mutazione che altera
la sequenza di DNA del gene stesso. Se questa mutazione ha luogo,
allora qualunque altro evento genetico che inattivi la rimanente
copia di Rd provoca l'innesco della trasformazone neoplastica.
Alcuni
autori hanno proposto, per quanto riguarda alcuni tumori maligni
che colpiscono l' infanzia, un meccanismo alternativo. Secondo
la loro opinione, l' evento che inattiva la prima copia di un
oncogene recessivo non sarebbe una vera e propria mutazione, ma
al contrario potrebbe essere un imprinting genomico, in grado
di disattivare i geni in maniera diversa nei maschi e nelle femmine..
In base a tale ipotesi, dunque, le cellule del rabdomiosarcoma
dovrebbero possedere sul cromosoma 11 sopravvissuto una copia
marcata, inattiva, del gene Rd. Inoltre nella maggior parte dei
casi di rabdomiosarcoma embrionale questo cromosoma proverrebbe
sempre dallo stesso genitore.
Sono
state in effetti trovate alcune prove che convalidano questa teoria.
La prima inattivazione nel rabdomiosarcoma embrionale, nel tumore
di Wilms e nell' osteosarcoma ha luogo quasi sempre in un oncogene
recessivo che si trova su un cromosoma paterno. I dati ottenuti
depongono in favore di un ruolo esercitato dall'imprinting genomico
nella genesi di questi tumori, ma prospettano anche un problema
teorico importante.
Il
problema nasce dal fatto che la maggior parte di coloro che hanno
studiato l'imprinting genomico, l'ha considerato come una conseguenza
della notevole differenza che esiste tra maschi e femmine, nella
fisiologia e nella biochimica della formazione dei gameti (uova
e spermatozoi). Nel corso della maggior parte della loro vita
da adulti, i maschi continuano a produrre miliardi di spermatozoi
a partire da una popolazione di cellule staminali che si dividono
ininterrottamente. Per contro, le femmine possiedono già
al momento della nascita tutte le cellule uovo che via via andranno
maturando nel corso del periodo riproduttivo. Se l' imprinting
differenziale riflettesse soltanto la dicotomia tra produzione
maschile e produzione femminile dei gameti, tutti i maschi dovrebbero
marcare i loro genomi in un certo modo e le femmine in un modo
diverso. Non potendo provare il contrario, abbiamo dato per scontato
che tutti gli appartenenti a uno dei due sessi marcassero i rispettivi
geni in maniera identica.
Ma
i dati che collegano i tumori maligni dell' infanzia a oncogeni
recessivi inattivi di origine paterna hanno smentito questo assunto.
Per esempio, se tutti i maschi marcassero e inattivassero il gene
Rd, l' incidenza del rabdomiosarcoma sarebbe molto elevata: una
sola alterazione genetica nel gene Rd ereditato dalla madre farebbe
trasformare alcune cellule di un individuo qualsiasi in cellule
tumorigene. Poiché i tumori di questo tipo sono rari,
(colpiscono approssimativamente un bambino su 20.000), è
improbabile che si erediti un oncogene recessivo inattivo dal
padre. Nei soggetti affetti da questo genere di malattia sembra
invece che accada esattamente questo.
Un'
affascinante spiegazione di questa differenza è che non
tutti i maschi marchino o inattivano gli stessi geni. Quale potrebbe
essere la causa di tali differenze tra individui? Se uno è
un genetista convinto, si rivolgerà sempre in primo luogo
alla genetica per ricevere risposta a interrogativi del genere.
Per una risposta di natura genetica devono esserci uno o
più geni responsabili dell' imprinting (da non confondersi
con quei geni che subiscono essi stessi l' imprinting ). In altre
parole, l' imprinting deve essere un processo che nasce dall'
attività di svariati geni che operano in maniera diversa
nei maschi e nelle femmine. I soggetti dello stesso sesso con
geni diversi che controllano l' imprinting avranno anche costellazioni
diverse di geni marcati. Per esempio, la maggior parte dei maschi
non disattiverà il gene Rd, ma lo faranno alcuni individui
con una copia aberrante di un gene che controlla l' imprinting.
L'
idea che l'espressione o la mancanza di espressione di un gene
possa essere controllata da altri geni non è nuova: descrive
un vecchio e ben noto fenomeno, definito "modificazione della
dominanza". Molti caratteri sono sensibili all'attività
di altri geni che si dice ne modifichino l'espressione. L'imprinting
genomico può essere visto come un caso speciale di modificazione
della dominanza. L'unica caratteristica insolita dei geni
modificatori che si presuppone controllino l'imprinting genomico
è quella di agire in maniera diversa nei maschi e nelle
femmine.
5) Espressione di materiale genetico transgenico in topi
transgenici
Una
osservazione importante riguarda il fatto che per circa un quarto
dei transgeni esaminati, l'espressione nelle generazioni successive
dipende dal sesso del genitore che trasmette il gene. Ad esempio
quando il gene è ereditato a partire da un topo transgenico
maschio, esso viene espresso nei tessuti appropriati. Tuttavia,
quando la figlia femmina del topo maschio transgenico trasmette
il gene alla propria prole, questa non lo esprime. A seguire,
i figli della prole maschile, esprimono il gene; invece, i figli
della prole femminile non lo esprimono. Questo fenomeno non sembra
collegato al sito di inserzione, alla grandezza del transgene,
al numero di copie incorporate; la non espressione pare associata
alla metilazione del transgene. Man mano che il gene passa da
una generazione all' altra, la sua metilazione viene invertita
a seconda dell' origine parentale. Per questo è stato suggerito
che la metilazione possa giocare un ruolo nella regolazione dell'
espressione dei geni coinvolti nell' imprinting.
Prese
insieme, queste cinque osservazioni suggeriscono con forza che
l' imprinting genomico (espressione differenziale del DNA derivato
dalla madre o dal padre) si verifichi davvero in alcune parti
del genoma dei mammiferi e ci si aspetta che svolga un ruolo essenziale
nelle patologie umane. Sembrerebbe che l' imprinting del DNA avvenga
normalmente durante la gametogenesi, in alcune regioni del genoma,
e che sia reversibile; vale a dire che non si tratta di una mutazione
o di un cambiamento permanente, ma piuttosto di una modificazione
che può essere eliminata quando le cellule germinali vengano
prodotte dalle generazioni successive.
Esaminare
un ipotetico pedigree di imprinting aiuta a visualizzare cosa
cercare negli studi familiari sull' imprinting genomico umano.
Un allele imprintabile viene trasmesso in maniera mendeliana,
ma la sua espressione sarà determinata dal sesso del genitore
che lo ha trasmesso.
Nell'imprinting
materno l'espressione fenotipica di un gene sia nella prole maschile
sia nella prole femminile, è influenzata dalla madre. Lo
" spegnimento " di tale gene si verifica se la prole
eredita il gene della madre, ma non quando lo stesso gene è
trasmesso dal padre di lei, dai fratelli, dai figli maschi. Quando
il figlio maschio (che non manifesta) trasmette il gene, la sua
prole lo esprimerà; invece, quando la figlia femmina (che
non manifesta) trasmette il gene, la sua prole non lo esprimerà.
Esattamente
il contrario avviene nell' imprinting paterno. Bisogna puntualizzare
che:
1 ) vengono osservati numeri uguali di maschi e femmine affetti
o che non manifestano ad ogni generazione, sia nell'imprinting
materno che paterno;
2) gli individui che trasmettono, ma non manifestano, rappresentano
la chiave per capire se un segmento cromosomico presenta imprinting
materno o paterno. Nell' imprinting materno è il maschio
il portatore che non esprime e trasmette ad una prole che manifesta;
nell' imprinting paterno, sono le femmine le portatrici che non
manifestano e trasmettono.
3 ) il pedigree di un gene imprintabile può sembrare autosomico
dominante, autosomico recessivo o ad ereditarietà multifattoriale,
a seconda della parte di famiglia che si osserva.
4) il pedigree è molto diverso da quello osservato nella
ereditarietà mitocondriale o citoplasmatica, in cui nessuno
dei figli o dei nipoti di un maschio può essere affetto,
mentre sono a rischio tutti i figli di una femmina.
Sapendo
del possibile effetto di imprinting sul fenotipo o sull' espressione,
in un qualsiasi disordine che manchi di uno schema di ereditarietà
chiaro, bisognerebbe esaminare il pedigree per scoprire prove
di imprinting. Molti disordini finora descritti come multifattoriali,
o come disordine che mostrano una grande variabilità di
espressione o come disordini con scarsa penetranza, sono dei candidati
particolarmente validi per questo tipo di effetto. Perciò
i pedigrees devono essere esaminati sistematicamente, in particolare
in quei disordini con schemi di ereditarietà insoliti.
Quando avviene l' imprinting?
L'
imprinting sembrerebbe un processo che possa modificare la sua
forma da un generazione alla successiva; vale a dire che non è
una mutazione permanente del DNA, ma piuttosto un' alterazione
temporanea della funzione di una parte del DNA (sebbene duri per
tutta la vita ). La differenza funzionale fra il genoma di origine
materna e quello di origine paterna, deve possedere una base molecolare
ben stabilita. Il meccanismo di imprinting implica:
- abolizione di ogni imprinting precedente;
- nuove modificazioni del genoma parentale nelle cellule germinali
di ciascun sesso;
- nuova etichettatura del cromosoma come paterno o come materno
(ciò potrebbe avvenire nello stesso momento in cui avvengono
delle nuove modificazioni del genoma);
- l'espressione fenotipica diversa tessuto-specifica del nuovo
imprinting nella prole.
L'
imprinting differenziale ereditato dai genitori deve essere cancellato
o inattivato nella linea di cellule germinali di ciascun individuo
affinchè venga introdotto un nuovo imprinting genomico.
In questo senso, l' imprinting autosomico ricorda il meccanismo
di inattivazione del cromosoma X. Le prove di cui si dispone suggeriscono
che almeno parte del processo che porta all'espressione differenziale
delle informazioni genetiche materne e paterne nell' embrione
e nel feto (e anche durante la vita) debba attuarsi durante la
gametogenesi. Il che significa che le nuove modificazioni potrebbero
avvenire durante la meiosi oppure durante gli stadi successivi
della maturazione dei gameti o alla fecondazione della cellula
uovo che diventerà zigote. D'altra parte è durante
il pachitene che i cromosomi omologhi si appaiano per tutta la
loro lunghezza e questo rappresenterebbe un momento logico perché
avvenga un qualche tipo di modificazione. Potrebbe essere un processo
correlato con lo stato fisico dei cromosomi (allungati o condensati,
metilati o non metilati) durante gli stadi della gametogenesi,
oppure potrebbe essere correlato a un processo attivo come il
centro di inattivazione della X.
Come avviene l' imprinting?
Il
lavoro compiuto sui topi transgenici suggerisce che l' espressione
differenziale del transgene imprintato sia associata alla metilazione.
Le modifiche del DNA attraverso la metilazione potrebbero fornire
una modalità per determinare se un particolare allele di
un gene sia in un certo momento inattivo. In generale, la metilazione
sembra essere un fenomeno secondario nella regolazione dei geni,
e potrebbe essere secondario anche in questo caso.
Effetti dell' imprinting.
Gli
effetti dell' imprinting appaiono in stadi precoci ed esplicano
la loro azione sulla crescita e sul comportamento. Anche gli incroci
tra specie diverse suggeriscono che l' imprinting possa avere
effetti sulla crescita di diverse aree del corpo; basti pensare
che nell' incrocio tra cavallo ed asino si osservano chiare differenze
a seconda se il cavallo è il padre ( mulo ) o la madre
(bardotto).
Conservazione dei segmenti imprintati?
Esperimenti
genetici effettuati con il topo hanno aiutato a stabilire una
mappa delle regioni del cromosoma imprintato. Potrebbe essere
che solo un piccolo numero di geni all' interno di ciascun segmento
sia in realtà soggetto ad imprinting. Si pone la questione,
nei topi e negli uomini, se ci sia conservazione delle regioni
imprintate oppure no. I confronti sono basati sulle recenti relazioni
a proposito della omologia o della sintenia fra cromosomi umani
e murini. Tali attribuzioni sono fin troppo abbozzate, tuttavia
rilevano una reale tendenza dei loci delle malattie imprintate
a sovrapporsi con i segmenti imprintati di quattro o cinque cromosomi
murini, che si sa essere imprintati. Non possono essere dedotte
conclusioni sulla base delle comparazioni, tuttavia queste suggeriscono
che veramente possa esistere una certa conservazione dei segmenti
imprintati tra i mammiferi.
Pubblicazioni chiave
La trasmissione materna ma non paterna di una mutazione
15q11-13 non da delezione provoca l'espressione fenotipica della
sindrome di Angelmann
J. Wagstaff, J.H.M. Knoll, K. A. Glatt, Y. Y. Shugart,
A. Sommer e M. Lalande, Nat.Genet 1 291-294 (1992)
La
sindrome di Angelman può derivare da delezioni del cromosoma
15q11-13 ereditato dalla madre, da disomia uniparentale paterna
del cromosoma 15, ma anche nel 40% dei pazienti, da una copia
del cromosoma 15 apparentemente intatta. Al contrario, la sindrome
di Prader-Willi può derivare da delezioni del cromosoma
15q11-13 ereditato dal padre o da disomia uniparentale materna
del cromosoma 15. Il fatto che le diverse origini parentali delle
delezioni del 15q provochino fenotipi diversi, ha portato a formulare
l'ipotesi secondo la quale uno o più geni localizzati in
questa regione mostrano espressione differenziale (imprinting
genetico).
Alcuni
autori hanno descritto una famiglia in cui i tre bambini affetti
da AS non mostrano una delezione citogenetica sul cromosoma 15.
L'analisi molecolare ha mostrato una delezione che coinvolge il
locus D15S10 in tutti e tre i bambini, così come nella
loro madre e nel loro nonno materno. Analisi successive hanno
mostrato che questa delezione sub-microscopica coinvolge anche
il locus che codifica la subunità B3 del recettore del
GABA (GABRB3 ). Studi recenti hanno evidenziato una regione critica
di sovrapposizione della delezione nel 15q11-13, che comprende
i loci D15S10 e GABRB3. Entrambi questi loci sono stati esclusi
dalla regione critica della delezione della PWS.
Il
fatto che la madre dei tre bambini affetti da AS abbia ereditato
la delezione che coinvolge i loci D15S10 e GABRB3 dal padre e
sia fenotipicamente normale (non mostra segni di PWS ), suggerisce
con forza che i loci genetici per AS e PWS, sebbene strettamente
associati, siano distinti. Il GABRB3 codifica per una subunità
(B3) del recettore per il GABA, che è un neurotrasmettitore
inibitorio. La presenza di questo gene nella regione critica della
AS, il cui fenotipo include: paresi, riso incontrollato, movimenti
anomali, è interessante, sebbene una correlazione diretta
tra questo gene e il fenotipo della AS non sia stata ancora stabilita. Un
secondo gene, che codifica per un' altra subunità del recettore
del GABA, la subunità A5, è stato recentemente mappato
al cromosoma 15q11-13, distalmente rispetto alla GABRB3, ed è
stato indicato come GABRA5.
Circa
un terzo dei pazienti affetti da AS non mostrano prove citogenetiche
di delezioni sul cromosoma 15, ma hanno ricevuto entrambi gli
alleli materni e paterni del cromosoma 15. Questo schema di non
delezione e' stato ritrovato in un certo numero di casi familiari
in cui e' presente piu' di un figlio con sindrome di AS; pertanto
il rischio di ricorrenza nei casi di AS senza delezione sembrerebbe
piu' alto rispetto ai casi deleti. Baraitser et al. hanno proposto
in questi casi una trasmissione di tipo autosomico recessivo.
Knoll et al. suggeriscono invece che questi pazienti potrebbero
aver ereditato un allele materno mutato e l' allele paterno normale,
di un locus nel 15q11-13 imprintato o alternativamente, che la
mutazione in un locus non localizzato in 15q11-13 potrebbe essere
la causa della comparsa del fenotipo AS. Infine, la disomia uniparentale
paterna per il cromosoma 15, spiega meno del 15 % dei casi di
AS; invece, nella PWS, la disomia uniparentale materna, sembra
spiegare tutti i casi che non presentano delezioni.
In
questa pubblicazione viene descritta una famiglia in cui tre sorelle
hanno avuto figli con AS senza delezione, una di loro con due
partners diversi. Per dimostrare che tutta la prole con AS ha
ereditato la stessa regione del nonno materno e che tutti gli
altri individui non affetti hanno ereditato la regione dalla nonna
materna, sono stati adoperati diversi polimorfismi del DNA all'
interno della regione 15q11-13. Una quarta sorella che non ha
avuto prole affetta da AS, ha ereditato gli altri alleli di questi
loci polimorfici del padre. Si suppone che la AS in questa famiglia
sia causata da una mutazione all' interno della regione 15q11-13,
la quale provoca il fenotipo AS se trasmessa da madre a figlio,
ma non lo provoca se trasmessa dal padre alle figlie.
Gli
individui affetti da AS ( fig.1) sono rappresentati da una coppia
di sorellastre da parte di madre e da due primi cugini materni.
Fig. 1
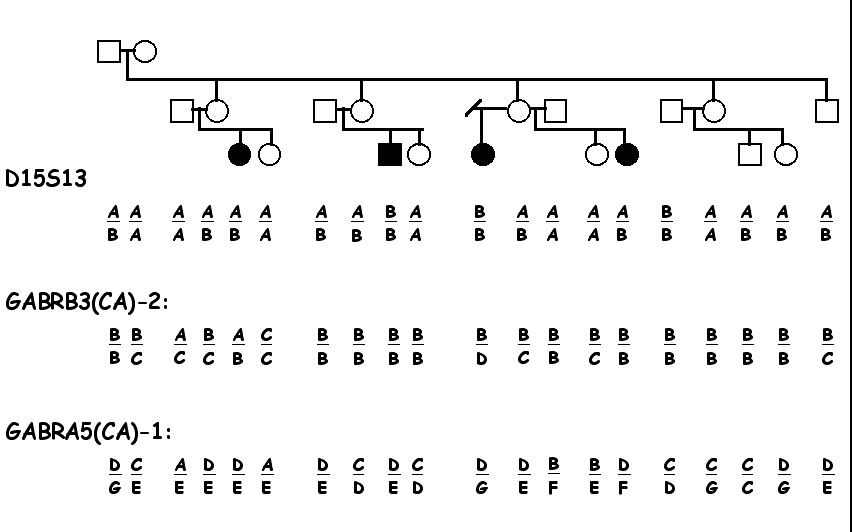
L'analisi
sia cromosomica che molecolare (tramite sonde polimorfiche della
regione) per tutti e quattro gli individui affetti non ha evidenziato
alcuna delezione del cromosoma 15. La sonda 189-1 (locus D15S13),
localizzata all' interno del segmento 15q11-13, prossimale alla
regione critica della AS, evidenzia polimorfismo Taq-1 a due alleli,
che fornisce importanti informazioni su questa famiglia (fig1).
Il nonno materno dei bambini affetti da AS è eterozigote
a questo locus (AB), mentre la nonna materna e'AA. Il nonno ha
trasmesso il suo allele B a ciascuna delle tre figlie che ha una
prole affetta da AS; invece, alla figlie che ha avuto due figli
non affetti, ha trasmesso l'allele A.. Tutti e quattro i bambini
affetti da AS, hanno ereditato l' allele B delle madri; i bambini
sani hanno ereditato l'allele A dalle madri (dalla nonna). Dunque,
in questa famiglia l'allele B al locus D15S13 viene ereditato
insieme ad un allele AS il quale, quando trasmesso dal padre ai
figli non produce effetti fenotipici, invece quando trasmesso
dalla madre ai figli produce il fenotipo AS. Inoltre, i genotipi
al locus D15S13 mostrano che per almeno due degli individui AS
( III-1 e III-7 ) è presente un allele materno, il che
esclude la possibilità di una disomia monoparentale paterna.
Ulteriori
prove, coerenti con l' ipotesi del nonno materno sia coinvolto
con l' espressione della AS, sono state fornite dal polimorfismo
GABRB3 (CA)-2 associato al locus GABRB3 anche se non completamente
informativo. Gli individui II-2 e II-5, madri di bambini affetti
da AS ereditano l'allele B dal padre e l' allele C dalla madre.
I figli con AS, III-1, III-5 e III-7, hanno ereditato l' allele
B dal nonno; quelli sani hanno ereditato l' allele C dalla nonna.
Un
altro polimorfismo che si è rivelato molto informativo
per questa famiglia è il GABRA5 (CA)-1 associato al locus
GABRA5. Le tre madri dei bambini affetti da AS, hanno ereditato
l' allele D dal padre mentre la quarta sorella eredita dal padre
l' allele G.I quattro bambini con AS hanno anch' essi ereditato
l'allele D del nonno, mentre i bambini sani hanno ereditato un
allele della nonna.
La
famiglia descritta rappresenta la prima prova diretta che la AS
senza delezione e senza disomia monoparentale, chiama in causa
una alterazione genetica della regione regione 15q11-13. La segregazione
di numerosi markers indica che tale alterazione è stata
trasmessa dal nonno materno (che è fenotipicamente normale
) a tre figlie femmine e ad un figlio maschio, tutti fenotipicamente
normali. Il fenotipo della AS si manifesta quando tale mutazione
viene trasmessa dalle figlie femmine alla propria prole, in coerenza
con l' ipotesi dell' imprinting. Inoltre il fatto che l'ipotetica
mutazione AS sia stata trasmessa da un maschio a quattro nipoti
sensa manifestazione di PWS nella generazione intermedia fornisce
un'ulteriore prova che i loci per PWS e AS siano distinti.
Il
modello di ereditarieta' di questa famiglia e' molto simile a
quello descritto per un allele mutato del fattore di crescita-II
insulino simile (IGF-II) nel topo. In questo caso solo la trasmissione
dell'allele paterno porta al fenotipo mutato (ritardo nella crescita).
La trsmissione materna non provoca nessun effetto fenotipico.
E' stato possibile nel topo provare questo tipo di trasmissione
grazie all'espressione limitata all'allele paterno in molti tessuti.
L'
attuale conoscenza della regione 15q11-13, coinvolta nella AS
e nella PWS, suggerisce che essa deve contenere almeno tre tipi
di loci (fig.2):
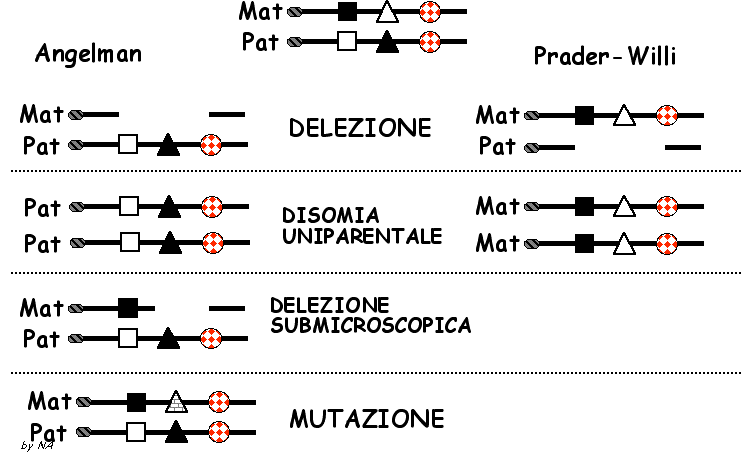
Fig. 2
1) geni di tipo I, preferenzialmente espressi sul cromosoma
materno, la cui delezione provoca la AS; tale espressione preferenziale
non deve necessariamente riguardare tutti i tessuti o tutti gli
stadi dello sviluppo. (Lo studio dell'espressione di IGF-II nel
topo indica che l'espressione del gene imprintato e' probabilmente
modificata sia nel tempo che nei tessuti nel corso dello sviluppo.)
2 ) geni di tipo II: sono espressi preferenzialmente sul cromosoma
paterno e la loro delezione causa la PWS.
3 ) geni di tipo III: sono probabilmente espressi in ugual modo
sia sul cromosoma materno sia su quello paterno. La presenza di
ipopigmentazione sia nei casi diAS che PWS dovuti a delezione,
suggerisce la presenza di un locus di tipo III in questa regione.
Studi di sintenia nel topo hanno evidenziato la presenza
di un locus p (occhio rosa) nella regione omologa alla
15q11-13 umana.
Secondo
il modello rappresentato nella fig.2, la AS risulta dalla mancanza
di espressione normale degli alleli materni dei geni di tipo I.
Questo potrebbe essere causato da una delezione materna, da una
disomia monoparentale paterna o da mutazioni sub-microscopiche
materne che distruggono questi geni.
La
PWS risulta dalla mancata espressione degli alleli paterni dei
geni di tipo II. Una recente analisi dei pazienti con PWS ha rilevato
che tutti gli individui clinicamente tipici hanno o una delezione
oppure una disomia monoparentale.
La
ragione della differenza con la AS, della quale sono riportati
un numero sostanziale di pazienti senza delezione, senza disomia
monoparentale, non è chiara. Forse nella PWS il bersaglio
della mutazione è molto più grande e probabilmente
coinvolge due geni non contigui, entrambi espressi preferenzialmente
sul cromosoma paterno.
Conversione dell'imprinting sul cromosoma 15 umano puo' coinvolgere
trascrittialternativi del gene SNRPN
Dittrich B.et al Nature Genetics 14 n.2 163-170 1996
Il
gene SNRPN ( Small Nuclear RibonucleoProtein N ) è espresso
solo dal cromosoma 15 paterno ed è localizzato in 15q11-13,
ricopre 25 kb di DNA genomico ed è costituito da dieci
esoni. La regione cromosomica 15q11-13 dell' uomo contiene almeno
altre quattro sequenze imprintate, che vengono espresse solo dal
cromosoma paterno; sono: i geni ZNF 127 e IPW, e due sequenze
scarsamente caratterizzate, la PAR5 e la PAR1.
Il
gene ZNF 127 mappa circa 1 Mb ed è centromerico rispetto
al gene SNRPN; i geni PAR5, IPW e PAR1, mappano, rispettivamente
50, 150 e 170 kb e sono telomerici rispetto alla isola CpG di
SNRPN.
I meccanismi che sottostanno all' imprinting genomico sono sconosciuti,
la metilazione specifica del DNA del genitore di origine sembra
giocare un ruolo importante nel regolare l' espressione del gene
imprintato. Infatti, è stata evidenziata una metilazione
diversa del DNA nel gene ZNF 127, nel gene D15S63, nell' isola
CpG e nell' esone 7 dell' SNRPN. La localizzazione cromosomica
e lo status dell' imprinting di questi geni, suggeriscono la possibilità
che siano coinvolti nell' eziologia della PWS. La causa della
PWS è la parziale mancanza del cromosoma 15; infatti,
può derivare da una delezione del cromosoma 15q11-13 paterno,
da una disomia monoparentale materna oppure da un difetto dell'
imprinting.
Esistono
prove indirette della presenza di almeno un gene, localizzato
sempre nel segmento 15q11-13, che però è espresso
dal cromosoma materno. Questo è il gene che viene
considerato essere coinvolto nella AS, la causa della AS è
la parziale mancanza del cromosoma 15 materno: può derivare
da una delezione del cromosoma 15q11-13 materno, da una disomia
monoparentale paterna, da una mutazione del gene AS o da un difetto
dell' imprinting. Si suppone che il gene AS mappi telomericamente
rispetto al gene SNRPN, ma non e' stato ancora indentificato.
L'
identificazione dei difetti dell' imprinting che portano alla
PWS e alla AS, offre l' opportunità unica di dissezionare
il meccanismo di imprinting nel segmento 15q11-13. Nel 50 % di
questi pazienti sono stati identificate delle microdelezioni,
che vengono ereditate, e che interessano una regione di poco meno
di 100 kb intorno e a monte dell' esone 1 del gene SNRPN. Queste
delezioni non esercitano sui geni PWS e AS soltanto un semplice
effetto di posizione, ma sembrano bloccare in cis la conversione
dell' imprinting paterno-materno durante la gametogenesi femminile,
o analogamente, materno-paterno durante la gametogenesi maschile.
Partendo
da questi risultati, gli Autori propongono che la struttura della
cromatina, la metilazione del DNA e l'espressione di un gene in
un dominio imprintato di 2 Mb all' interno della regione 15q11-13,
siano regolati in cis da un centro di imprinting ( IC ), localizzato
tra i geni D15S63 e SNRPN.
In
questo lavoro viene dimostrato che questa regione codifica trascritti
alternativi di SNRPN e che vi sono presenti mutazioni intrageniche
in pazienti AS e PWS originati da fallimento della conversione
dell'imprinting; e propongono un meccanismo di conversione dell'imprinting
durante la gametogenesi.
Per
isolare le sequenze espresse dal centro di imprinting e per selezionare
cloni di cDNA fetale umano dal cervello, dal fegato e dal muscolo
umano adulto, è stato usato un contiguo fagico di 160 kb
compreso fra D15S63 (PW71) e SNRPN. Un clone cDNA del cervello,
il bd48 ( 392bp ), si è ibridato a parecchi frammenti EcoRI
del contiguo fagico. Confrontando la sequenza del bd48 con la
sequenza di questi frammenti EcoRI, è stato osservato che
l' inserto contiene tre esoni nuovi (denominati: BD1B, BD2, BD3)
a 27 bp dall' estremità 5' dell' esone 2 del gene SNRPN
(fig.3). La distanza genomica ricoperta dal bd48 è di circa
150 kb e la direzione della trascrizione è dal centromero
verso il telomero.
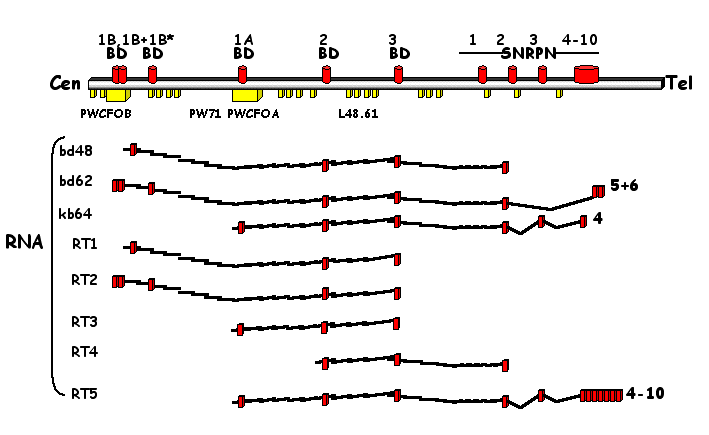
Fig.3 Mappa fisica della regione e struttura dei cDNA
Si
è visto che il clone bd48 si sovrappone ad altri due cloni
selezionati dall'RNA del cervello (bd62 e kb74, fig.3). Il clone
bd62 contiene l'esone BD1B più 27 bp addizionali (esone
BD1B'), l'esone BD1B*, l'esone BD2 e l'esone BD3, così
come l'esone 2, 5 e parte del 6 dell' SNRPN. Il clone Kb74
contiene gli esoni BD1A, BD2 e BD3, come pure gli esoni 2, 3 e
4 dell' SNRPN. In tutti i trascritti BD manca l'esone 1 del gene
SNRPN, ma e' presente l'esone 2. Tutte le giunzioni esone/introne
corrispondono alla sequenza consensus del sito di splicing, e
gli esoni BD non contengono una lunga ORF (open reading frame).
La scoperta di esoni alternativi nei tre cloni di cDNA, ha fornito
prove che fanno sospettare uno splicing alternativo o siti alternativi
di inizio della trascrizione. Per analizzare ciò con maggiore
dettaglio, è stata effettuata una ( RT )-PCR su RNA di
cervello fetale umano.
Usando
primers specifici per BD1B e BD3, sono state ottenute due bande
di 335 bp e 225 bp ( RT2, RT1; fig.3): queste bande sono state
purificate, riamplificate e sequenziate. Il frammento da 335 bp
contiene gli esoni BD1B, BD1B*, BD2 e BD3; invece, nel frammento
da 225 bp manca l' esone BD1B*. Si assume, quindi che l' esone
BD1B* sia alternativo. Successivamente, adoperando un primer per
l'esone BD2 ( che non era presente in RT1 e RT2 ), si ottiene
un prodotto PCR di 230 bp ( RT3 ). Il sequenziamento ha rivelato
la presenza in RT3 degli esoni BD1A, BD2 e BD3. Questi dati indicano
che l'esone BD1A non è alternativo nei trascritti che iniziano
con l'esone BD1B, ma è un esone 5' alternativo.
Per
verificare che i trascritti BD abbiano esoni SNRPN all'estremità
3', è stata effettuata una (RT)PCR con l'RNA di cervello
fetale umano con un primer che si lega all'esone BD2 e con altri
primer che si legano all'esone 2 e 10 del SNRPN. Nel primo caso
è stato osservato un prodotto di 245 bp ( RT4, fig.3);
il sequenziamento di tale prodotto, riamplificato e purificato,
ha confermato che l' esone 2 dell' SNRPN si trova a valle dell'
esone BD3. Nel secondo caso (RT5, fig3) non è stato ottenuto
un prodotto visibile, ma ibridazioni successive con una sonda
specifica per BD ed una sonda specifica per SNRPN, hanno evidenziato
due bande di 1,3 kb e di 0,6 kb. Mentre risulta sconosciuta la
natura del prodotto di 0,6 kb, 1,3 kb e' la misura che ci si attendeva
da un prodotto (RT)- PCR che contenga l'esone BD3 legato agli
esoni 2-10 di SNRPN.
Per
definire le estremità 5' e 3' dei nuovi trascritti è
stata effettuata la RACE: Rapid Amplification of cDNA Ends ( amplificazione
rapida delle estremità del cDNA ). Usando RNA di cervello
fetale umano e primers che si legano all'esone BD2, è stato
ottenuto un prodotto RACE dell'estremità 3', di 170 bp.
L'analisi di sequenza ha evidenziato che tale prodotto contiene
parte di BD2 e la porzione 3' dell'esone 10 di SNRPN, seguita
da una coda di poli (A).
Facendo
la 5' RACE con primers che si legano all' esone BD2, si può
estendere la sequenza dell' esone BD1A di 78 bp; non è
stato ottenuto nessun prodotto RACE che si estendesse oltre l'
esone BD1B.
L'esone
BD1A mappa vicino al sito CfoI sensibile alla metilazione,
evidenziato da PW71 (D15S63). La sequenza di DNA che comprende
questo sito e il BD1A (PWCFOA) condivide l' 82 % di omologia con
la sequenza di DNA che comprende il BD1B (PWCFOB). L'analisi del
pattern di metilazione del sito CfoI in PWCFOB eseguita
su pazienti PWS, AS e controlli normali ha messo in evidenza la
presenza di differenze (due bande di intensita' analoga nei normali,
una delle due bande piu' debole nei PWS, assenza dell'altra banda
nei pazienti AS). Da questi dati si puo' dedurre che il sito CfoI
materno e' metilato nella maggior parte delle cellule, mentre
il paterno e' sottometilato tanto in PWCFOA che in PWCFOB. In
sintesi si puo' dire che questi due trascritti vengono iniziati
a livello di due siti omologhi (PWCFOA e PWCFOB) metilati in modo
differenziale, che sono soggetti a splicing alternativo e che
hanno gli esoni di SNRPN al loro 3'.
L'espressione
del trascritto BD è stata determinata utilizzando delle
sonde, prive di sequenze SNRPN, che vengono ibridate in un Northern
blot con diversi tessuti umani. Nel cervello fetale umano è
stato evidenziato un segnale di circa 1,7 kb, lo stesso in polmoni,
fegato e reni fetali; nel cuore, nel cervello, nella prostata,
nei testicoli e nelle ovaie, nell' intestino, nel colon, dell'
individuo adulto. D' altra parte, non è stato ottenuto
nessun segnale nell' adulto, a livello di placenta, polmoni, fegato,
muscolo scheletrico, rene, pancreas, milza, timo, sangue periferico.
L'espressione più forte è stata osservata nel cervello
e nel cuore. La grandezza del trascritto corrisponde alla misura
attesa dallo splicing degli esoni BD (365- 489 bp) con gli esoni
2-10 dell' SNRPN (1251 bp).
Per
determinare se nei pazienti con PWS e AS i geni BD vengano trascritti
sia dall'allele materno che paterno oppure da uno soltanto, è
stato analizzato RNA da una library di cervello di individui normali
(i trascritti BD non vengono espressi nei linfoblasti e quindi
non si poteva esaminare direttamente il DNA dei pazienti [n.d.t.])
e utilizzato un polimorfismo BamHI per l'esone BD1B*. Con il Southern
blot sono stati osservati due alleli di 10,3 kb e 7,9 kb. Nella
sequenza PWCFOB, che non è metilata sul cromosoma paterno
e metilata su quello materno, il sito BamHI mappa telomericamente
rispetto al sito CfoI. Un individuo, indicato come S367/95, dei
sei analizzati, è risultato eterozigote per questo polimorfismo
BamHI, nell' esone BD1B*. La banda di 10,3 kb che rappresenta
l'allele paterno viene completamente tagliata da CfoI; invece
la banda di 7,9 kb è parzialmente resistente al taglio
del CfoI e quindi rappresenta l' allele materno. Questi risultati
dimostrano che il cromosoma paterno manca del sito variabile BamHI
nell' esone BD1B*, che è al contrario presente sul cromosoma
materno.
Quando l' RNA del cervello di tale individuo è stato inversamente
trascritto e amplificato con primers specifici per gli esoni BD1B
e BD3, sono state ottenute due bande di 335 bp e di 225 bp (RT2,
RT1;
fig.3). Il frammento di 335 bp, che contiene l' esone BD1B*, è
stato purificato e riamplificato: tutte le molecole mancano del
sito BamHI. Ciò significa che l'esone BD1B* è trascritto
solo dal cromosoma paterno.
Identificazione delle mutazioni intrageniche
Sono
state studiate tre famiglie con PWS in cui era stata osservata
una microdelezione dell'esone 1 dell' SNRPN che blocca la conversione
dell' imprinting materno-paterno, in contrasto con altre famiglie
AS in cui la microdelezione blocca la conversione paterno-materno.
In cinque delle sei famiglie con AS di questo tipo e' presente
una delezione dell' esone BD3 (frammento L48.6I [fig4.]), mentre
la sesta indicata con AS-H, non mostra alcuna delezione dell'esone
BD3, tuttavia presenta una delezione di 6 kb solo a 288 bp a valle
del BD3. E' possibile che questa delezione interferisca con lo
splicing o la trascrizione del trascritto BD, ma non e' possibile
testare l'ipotesi per la mancata espressione di BD in tessuti
disponibili dai pazienti AS.
Utilizzando
SSCP e l'analisi di sequenza sono stati analizzati gli esoni BD2
e BD3 in 6 pazienti AS di famiglie in cui non era presente microdelezione.
In una (AS-C2,fig.4) e' stata trovata una mutazione puntiforme
che altera il donor site di splicing, tale mutazione non e' stata
ritrovata in nessuno dei 44 contralli testati ne nel padre, la
madre non era reperibile per l'analisi.
Nelle
famiglie con PWS il paziente aveva la delezione sul cromosoma
paterno, mentre il padre e la nonna paterna l'avevano sul cromosoma
di origine materna. Nelle famiglie con AS, il paziente aveva la
delezione sul cromosoma materno, mentre la madre l'aveva sul cromosoma
paterno.
DISCUSSIONE
Identificazione del trascritto dal centro di imprinting
Il
cromosoma 15 umano contiene un centro di imprinting (IC) che regola
in cis attraverso un dominio cromosomico di 2Mb all' interno
del cromosoma 15q11-13, la struttura cromatinica, la metilazione
del DNA e l' espressione dei geni. La presenza dell' IC
era stata dedotta dall'analisi genetica di famiglie con PWS e
con AS che presentano un difetto nel processo di imprinting, e
hanno mutazioni in questa unita' trascrizionale.
Dallo
studio condotto si puo' dedurre che la regione IC codifica dei
trascritti alternativi del gene SNRPN. Cinque
delle famiglie AS con mutazione dell' imprinting studiate presentano
delezioni che interessano almeno un esone BD; in particolare (fig.4):
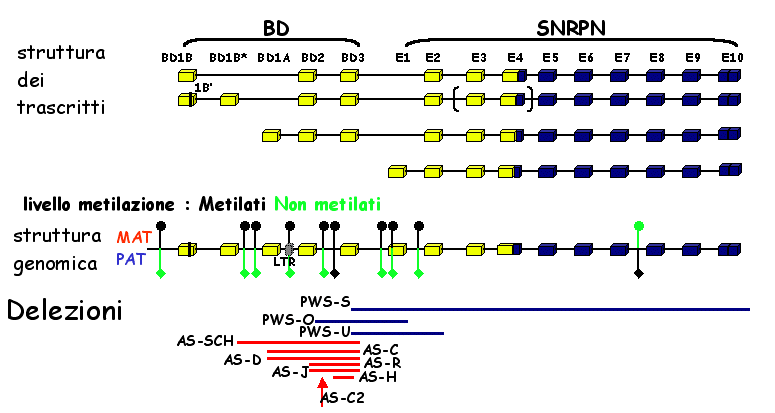
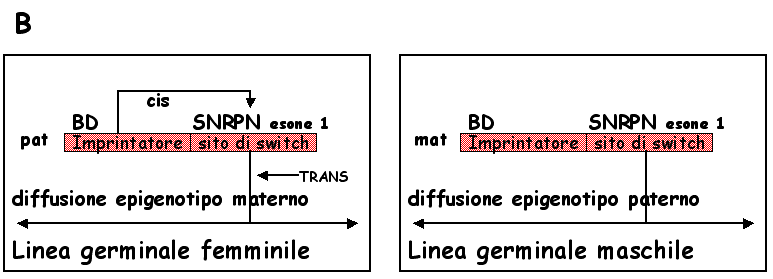
Fig.4. Struttura e funzione del centro di imprinting.
A) Sruttura dei trascritti e organizzazione genomica dell'unita'
di trascrizione di SNRPN. I quadrati indicano gli
esoni, l'open reading frame di SNRPN e' colorata
di nero; il piccolo quadrato grigio indica un'isola LRT; gli esoni
3 e 4 del secondo trascritto sono in parantesi perche' risultano
assenti nel clone bd62 (cfr.fig3). La localizzazione e l'estenzione
delle delezioni che bloccano la conversione dell'imprinting sono
indicate come linee sotto la mappa, la fraccia indica la mutazione
puntiforme trovata nella famiglia AS-C2. I cerchietti neri indicano
i siti HpaII, CfpI o Not! metilati, i cerchietti bianchi
quelli non metilati, quelli a meta' isiti metilati nel 50% delle
cellule. B). modello per la conversione dell'imprinting.
Due famiglie ( AS-R, AS-J ) hanno delezioni solo nell'esone BD3,
altre due famiglie (AS-C e AS-D) mostrano delezioni per BD2 e
BD3, una famiglia ( AS-SCH ) mostra delezione per BD1A, BD2 e
BD3. Un paziente ha una mutazione puntiforme (AS-C2) nel donor
site, mentre in un'altro (AS-H) la delezione distale a BD3 puo'
interferire con l'espressione del trascritto. In tutte le famiglie
con PWS (3) l'esone 1 di SNRPN e' deleto.
Sebbene
questi dati suggeriscano un ruolo dei trascritti alternativi dell'
SNRPN nel processo di conversione dell' imprinting, non si è
ancora in grado di calcolare la possibilità che le mutazioni
colpiscano degli elementi regolatori o dei trascritti compenetrati,
ma non correlati, con il complesso di trascrizione SNRPN.
Natura dei trascritti
I
trascritti BD hanno due siti di inizio alternativi e sono soggetti
a splicing alternativi. I due esoni alternativi al 5' (BD1A e
BD1B)sono parte di di due sequenze di DNA omologhe (PWCFOA e PWCFOB),
che sono metilate sul cromosoma materno e sottometilate nel paterno.
Questo e' vero per la maggior parte delle isole GpC con metelazione
alternativa presenti nella regione IC (fig.4). E' probabile che
la metilazione sia coinvolta nella regolazione dell'espressione
dei trascritti della regione. Inaspettatmente i due esoni BD sono
esoni 5' alternativi del gene SNRPN; essi contengono delle ORF
brevi che non corrispondono alla sequenza Kozak di consenso per
i siti di inizio. La reading frames dell' SNRPN comincia all'
esone 4 e non è influenzata dallo splicing degli esoni
BD all'esone 2. Comunque il clone bd62 e' privo degli esoni 3
e 4 del gene SNRPN e quindi del sito di inizio per la proteina
SNRPN. Questi dati suggeriscono che i trascritti BD siano privi
di un potenziale codificante. In confronto a SRNPN i trascritti
BD sono espressi in meno tessuti e ad un livello inferiore. I
trascritti BD sono stati principalmente trovati nel cervello,
cuore, testicoli e ovaio ed e' stato dimostrato (usando il polimorfismo
BamH1 e lo stato di metilazione delle regioni di inizio PWCFOA
e PWCFOAbB [n.d.t.] )che derivano dal solo cromosoma paterno.
Un modello per la conversione dell'imprinting.
E'
stato mostrato che in parecchie famiglie AS delezioni e mutazioni
puntiformi degli esoni BD sono associate ad un blocco della conversione
dell' imprinting paterno-materno; mentre le delezioni dell' esone
1 di SNRPN sono associate ad un blocco dello conversione dell'
imprinting materno-paterno, in parecchie famiglie PWS. Le osservazioni
compiute suggeriscono che il centro di imprinting abbia una struttura
bipartita. Un qualsiasi modello che provi a
spiegare la funzione del centro di imprinting deve considerare
questa struttura, l'espressione paterna dei trascritti che si
estendono in questa regione (assumendo che siano espressi nelle
cellule della linea germinale ) e l' effetto delle mutazioni dell'
IC nell' ambito del cromosoma 15q11-13. Il seguente modello e'
compatibile con i dati ottenuti, ma bisogna sottolineare che non
intende spiegare come un singolo gene sia regolato dall'epigenotipo
materno o paterno del dominio cromosomico imprintato, ma come
possa avvenire la conversione degli epigenotipi paterno e materno
durante la gametogenesi.
In
questo modello, il centro dell' imprinting consiste di un imprintatore
e di un sito di inizio della conversione dell'imprinting ( fig.4B).
L' imprintatore codifica il trascritto BD; è espresso soltanto
dal cromosoma paterno e agisce in cis sul sito di inizio della
conversione (esone 1 dell' SNRPN o un sito vicino), forse inducendo
un cambiamento locale nella struttura della cromatina. Questo
cambiamento potrebbe essere provocato dalla trascrizione oppure
dall'interazione dei trascritti BD con il DNA. La mutazione di
splicing trovata nella famiglia AS-C2 suggerisce che per il funzionamento
sia importante il trascritto in se piuttosto che il processo di
trascrizione.
Nella
linea germinale XX un fattore che agisce in trans, specifico per
le cellule della linea germinale femminile, è coinvolto
nel completare lo conversione e/o nell' iniziare la diffusione
bidirezionale dell' imprinting materno sul cromosoma derivato
dal padre e rende silente l' attività dell' imprintatore.
Nella linea germinale XY, in assenza del fattore che agisce in
trans, l' imprinting materno sul cromosoma derivato dalla madre,
viene cancellato a partire dal sito di inizio della conversione
ed inizia la diffusione dell' imprinting paterno. La conversione
materno-paterno potrebbe avvenire per " default " oppure
implicare altri fattori.
Sebbene
ci siano delle valide indicazioni che il trascritto BD sia quello
dell' imprintatore che avvia l' imprinting paterno-materno durante
la gametogenesi femminile, il ruolo della regione deleta dell'
esone 1 del gene SNRPN nelle famiglie PWS con mutazione dell'imprinting
è meno chiaro. Poiché il gene SNRPN è inattivo
sul cromosoma materno, la trascrizione dello stesso con scarsa
probabilità avvia lo conversione dell' imprinting materno-paterno
durante la gametogenesi maschile. Una possibilità è
che l' esone 1 dell' SNRPN rappresenti il sito di inizio dello
conversione dell' imprinting.
Sulla
base di dati che riguardano la metilazione e la replicazione del
DNA, è stato proposto che la copia paterna imprintata del
dominio all' interno del 15q11-13, potrebbe avere una struttura
cromatinica simile all' eucromatina, dalla quale i geni PWS vengono
trascritti, mentre la copia materna potrebbe avere una struttura
cromatinica simile all' eterocromatina, dalla quale il/i gene/i
della AS è/sono trascritto/i. E' possibile
che il sito di inizio dello conversione rappresenti un centro
di nucleazione per la condensazione e la decondensazione dell'
eterocromatina, reso accessibile dal trascritto dell'IC. Questo
meccanismo e' stato proposto per l'azione di Xist nell'inattivazione
del cromosoma X. Nei mammiferi non sono stati ancora identificati
i siti di induzione dell' eterocromatina, ma questi sono ben conosciuti
in Drosophila. In questa specie, il mantenimento degli
schemi di espressione dei geni attivi e inattivi durante lo sviluppo,
implica strutture cromatiniche più complesse.
Il
modello spiega perché le microdelezioni osservate nelle
famiglie con PWS e AS con mutazione dell' imprinting bloccano
lo conversione dell' imprinting. Nelle famiglie PWS la conversione
dell' imprinting materno-paterno è bloccata a causa di
una mutazione nel sito di inizio dello conversione . Nelle famiglie
AS la conversione dell' imprinting paterno-materno è bloccato
a causa di una mutazione dell' imprintatore .
Il modello implica che una mutazione
dell' imprintatore blocchi la conversione paterno-materno, ma
non quello materno-paterno. Ciò è esattamente quello
che è stato riscontrato nella famiglia AS-C con sindrome
di Angelman. Questi risultati vengono interpretati nel modo seguente:
il nonno, portatore della delezione sul cromosoma di origine materna,
è in grado di effettuare la conversione materno-paterno
perché quest' ultima non coinvolge i geni BD. Sua
figlia (madre del paziente AS ), che ha ereditato la delezione
dal padre, non è in grado di effettuare lo conversione
paterno-materno, poiché è necessaria l'attività
BD (fig.5). A prima vista può sembrare sorprendente che
l' imprinting materno dipenda dall' attività del gene paterno
(espressione BD ). Comunque, per spiegare questo si possono immaginare
diverse ipotesi.
Innanzitutto,
per iniziare il processo di conversione paterno-materno dell'
imprinting, è necessaria l' espressione del gene. Come
accade durante l' inattivazione del cromosoma X, il cromosoma
che esprime il trascritto del centro di inattivazione viene spostato
da uno stato simile all' eucromatina ad uno stato simile all'
eterocromatina. In secondo luogo, il meccanismo di conversione
proposto si regola da sé: l'espandersi dell' imprinting
materno rende silente l'attività dell' imprintatore. Al
contrario, l' espandersi dell' imprinting paterno riattiva l'
imprintatore. Questo è importante perché diversamente
dall'inattivazione del cromosoma X, nei tessuti embrionali femminili
la conversione non e' casuale. Infine, il meccanismo di autoregolazione
fornisce una spiegazione circa l' evoluzione dell' imprinting
nel cromosoma 15q11-13. L'epigenotipo paterno sembra simile all'
epigenotipo di altre regione cromosomiche non imprintate. L' epigenotipo
materno potrebbe essersi sviluppato da questo epigenotipo acquisendo
gli esoni BD e i fattori trans agenti.
Questo
modello è basato su dati genetici ottenuti dal mappaggio
dei geni e dall' analisi delle mutazioni in famiglie AS e PWS
che presentano un errore di conversione dell' imprinting. Di conseguenza
tale modello non è in grado di fare previsioni a proposito
del destino dell'epigenotipo paterno sul cromosoma paterno nella
linea germinale maschile e dell'epigenotipo materno sul cromosoma
materno nella linea germinale femminile. Per esempio, non è
dato sapere se l' imprinting su questi cromosomi sia mantenuto
o sia rimosso e poi ristabilito. Questo problema potrebbe essere
investigato in pazienti che hanno AS o PWS dovuta ad un errore
di mantenimento dell' imprinting; pazienti di questo tipo non
sono stati ancora riportati.
Commento di Anne C. Fergusson- Smith
Nature Genetics 14 n.2 119-123 1996
Il
lavoro di Dittrich e dei suoi collaboratori fornisce una visuale
più ampia del meccanismo di imprinting. Ricapitolando brevemente,
l' imprinting implica l' acquisizione di segnali specifici della
linea germinale, conferiti sul DNA o sulla cromatina, al momento
della fecondazione, o immediatamente dopo, inducendo un'attività
differenziale dei due alleli parentali. E' importante che l'imprinting
venga cancellato nella linea germinale affinchè possano
essere acquisiti nuovi imprinting specifici dei genitori. Errori
nella linea germinale, che bloccano il cancellamento dell' imprinting,
impedirebbero agli omologhi di acquisire l'epigenotipo materno
o paterno e di influenzare l' attività dei geni imprintati.
Il
lavoro di ricerca di Dittrich e altri, sull' analisi delle mutazioni
PWS / AS che colpiscono il processo di imprinting della linea
germinale, ha reso possibile l' identificazione di una regione,
che è stata definita centro di imprinting (IC), e che regola
in cis l' imprinting appropriato di un dominio di 2 Mb, associato
ad entrambi le sindromi, che contiene parecchi geni imprintati,
compreso il gene SNRPN espresso paternalmente. Le mutazioni dell'IC
ereditate dal padre impediscono alla linea germinale XY di compiere
lo conversione materno-paterno e provocano una ritenzione dell'
epigenotipo materno (fig.6 generazione II e III).
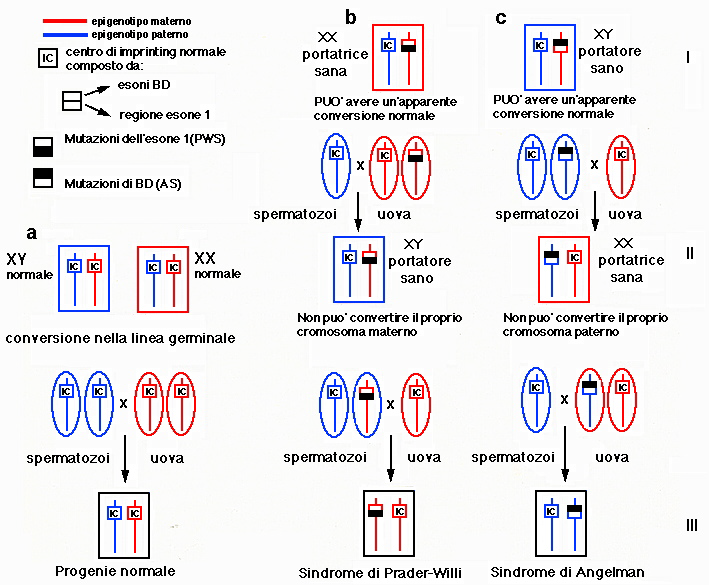
Fig.6. Rappresentazione schematica dell'azione del centro
di imprinting IC nelle linee germinali normali (a), nel caso di
mutazioni PWS (b), nel caso di mutazioni AS (c). Le generazioni
sono indicate sulla destra.
La
prole che eredita questo cromosoma non " convertito "
dal padre, avrà due epigenotipi materni e di conseguenza
la PWS. Nella linea germinale XX le mutazioni dell' IC impediscono
al cromosoma paterno di acquisire un epigenotipo materno e quindi
la prole ha due epigenotipi paterni e di conseguenza la AS (fig.6
generazioni II e III ). I dati ottenuti da Dittrich e altri mostrano
con chiarezza che gli esoni BD sono richiesti per una conversione
paterno-materno ( linea XX ) e l'esone 1 di SNRPN è richiesto
per lo conversione materno-paterno ( linea XY ), a suggerire una
struttura bipartita dell' IC.
Con
questo scenario in mente, piuttosto complesso, questi autori propongono
un modello per spiegare come due diverse regioni permettano la
acquisizione di un epigenotipo materno o paterno a seconda della
linea germinale attraverso cui passano. Secondo questo modello,
che assume l'espressione solo paterna di SNRPN, nella linea germinale
XX per attivare il cromosoma paterno è richiesta l'attività
di un imprintatore (codificato dai geni BD) che interagisce con
l'esone 1 di SNRPN; ciò rende il cromosoma accessibile
ad un fattore sesso-specifico che agisce in trans responsabile
dell' acquisizione dell' epigenotipo materno. In questo modello
il cromosoma ereditato dalla madre mantiene l'epigenotiopo materno.
Nei soggetti con AS, poiché gli esoni BD sono mutanti,
l' imprintatore non opera sul cromosoma paterno e di conseguenza
il fattore che agisce in trans non ha accesso su tale cromosoma
e la conversione paterno-materno è bloccata.
Nella
linea germinale XY in assenza del fattore che agisce in trans
l'epigenotipo paterno e' mantenuto e che anche in assenza di attivita'
di BD, il cromosoma materno acquisisce l' epigenotipo paterno
per "default". Nei soggetti con PWS la mutazione dell'esone
1 di SNRPN non permetterebbe al cromosoma materno l'acquisizione
dell'epigenotipo paterno, suggerendo un coinvolgimento dell'esone
1 nel mantenimento di default".
Un'
altra possibilità è che in PWS non vi sia il cancellamento
dell'imprinting e l'esone 1 dell' SNRPN sia coinvolto anche nella
cancellazione. Infatti nel topo esistono prove di un meccanismo
che implica sia la cancellazione sia il ristabilirsi di nuovi
imprinting nella linea germinale: il gene Snrpn murino
è espresso biallelicamente nelle cellule germinali. Sarebbe
possibile ipotizzare un modello a due steps ( fig.7)
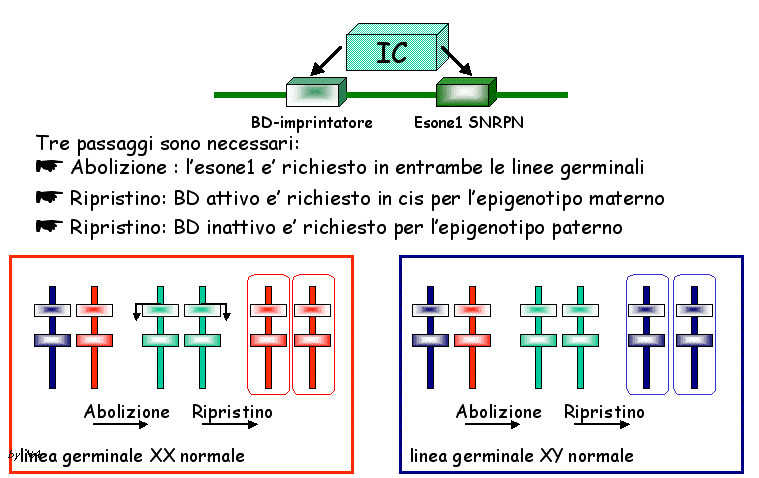
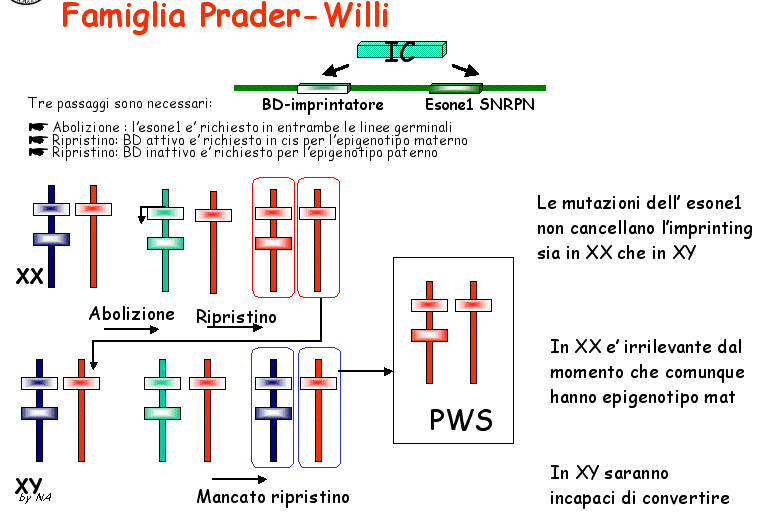
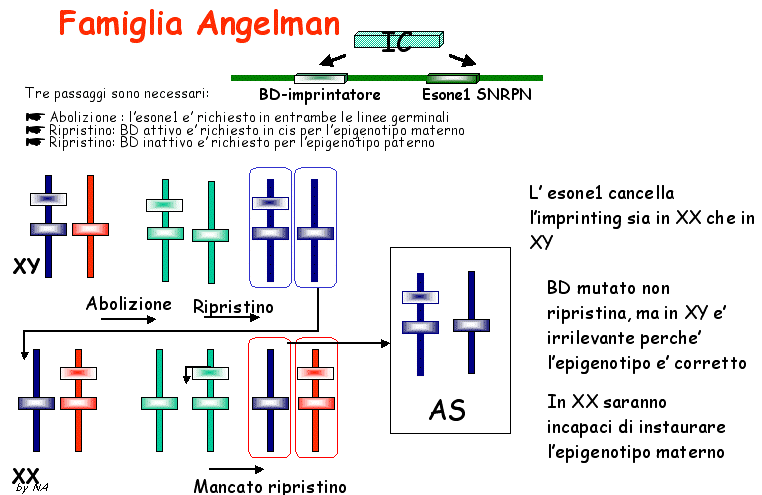
Fig.7. Ipotetico modello a due fasi: 1) Abolizione e 2)
ripristino : l'esone1 viene coinvolto nell'abolizione dell'imprinting
piuttosto che nella diffusione dell'imprinting.
secondo il quale l' esone 1 di SNRPN deve cancellare il precedente
imprinting ed in un secondo momento deve intervenire l' imprintatore,
il quale nella linea germinale XX interagisce con l' esone 1 per
l' acquisizione dell' epigenotipo materno. Nella linea germinale
XY, per l' acquisizione dell'epigenotipo paterno è necessaria
l'assenza dei BD. Nella PWS l' esone 1 è mutato per cui
non può cancellare l'imprinting esistente, sia nella linea
germinale femminile che maschile. Tuttavia, nella linea germinale
XX le mutazioni ereditate dalla madre non hanno importanza perché
avranno un epigenotipo materno. Nella linea germinale XY ciò
comporta l' impossibilità di conversione.
Il centro di imprinting sul cromosoma 15 si supponeva unico ma
di recente la scoperta di variazioni dell' imprinting nella Sindrome
di Beckwith-Wiedemann ( BWS ), suggerisce che possa esistere un
centro di imprinting anche sul cromosoma 11.