Sintesi di: Towards a molecular understanding of Prader-Willi and Angelman syndromes
Mellissa R. W. Mann, Marisa S. Bartolomei
Human Molecular Genetics 1999 Vol 8 n. 10 Pages 1867-1873
In mammals, both parents contribute equal genetic information
to their offspring. This normal diploid complement means that
most autosomal genes will be expressed from both the maternal
and paternal alleles. A small group of genes defies this normal
Mendelian mode of inheritance. Instead, imprinted genes are expressed
from only one of the two alleles in a parent-of-origin-dependent
manner. These genes are designated as imprinted since they retain
the parental identity they acquired during gametogenesis. The
regulation of imprinted genes is orchestrated by an epigenetic
modification to DNA. As such, imprinted genes are not only susceptible
to changes in the genetic sequence but also to disruptions in
the epigenetic program that controls these genes.
The mechanisms controlling genomic imprinting are likely to be
complex and at present are poorly understood (1-4). What is clear
is that deviation from appropriate parent-of-origin-dependent
expression may have dire consequences for the organism. Aberrant
imprinted gene expression has now been determined to be the cause
of a number of human diseases, including Prader-Willi syndrome
(PWS) and Angelman syndrome (AS), emphasizing the importance of
correct parental-specific expression of imprinted genes. PWS and
AS are two classic examples of imprinting in humans (5, 6). .
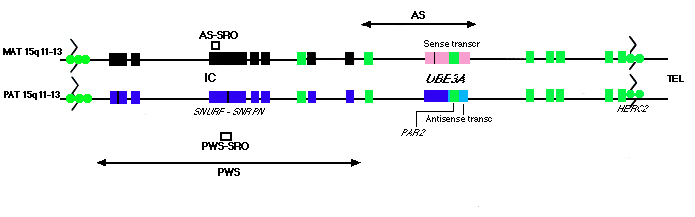
GENETIC ETIOLOGY OF PWS AND AS
PWS and AS are clinically distinct neurological disorders.
Several molecular mechanisms have been identified that lead to
PWS and AS, in all cases, loss of expression of at least one paternally
expressed or one maternally expressed gene, respectively, at 15q11-q13
is the causative event in the development of these syndromes.
The most common molecular defect giving rise to these syndromes
is a large chromosomal deletion (~4 Mb) that includes a large
cluster of imprinted genes (2-3 Mb) and a non-imprinted domain
(1-2 Mb) (9, 10). Paternal inheritance of the deletion results
in PWS while maternal inheritance produces AS. In addition to
the same rate of occurrence (~70%), deletions in PWS and AS occupy
the same cytogenetic position, 15q11-q13. Molecular analysis of
the breakpoint ends indicates that the vast majority of deletions
cluster at distinct sites (9, 11). Two breakpoint clusters have
been mapped centromeric to ZNF127, with the more proximal breakpoint
accounting for ~65% of deletions (12). The distal breakpoint cluster
has been mapped telomeric to the P locus. The inherent instability
of 15q11-q13 may be attributed to an expressed low copy repeat
sequence which is located in the vicinity of the breakpoint clusters
(12, 13). This sequence appears to be housed within the HERC2
gene, which encodes a gigantic HECT (homologous to E6-AP C-terminus)
and RCC1 (regulator of chromatin condensation) domain protein
and is located telomeric to P (13). At least seven expressed pseudogenes
arising from genomic duplication of HERC2 are present in the human
genome, including two copies that are located adjacent to the
HERC2 locus at 15q13, three pseudogenes that were translocated
to 15q11 and two pseudogenes that reside at 16p11. 2.
| Figura1. Genetic map of human chromosome 15q11-q13. The translocation breakpoint clusters (zigzag line) associated with chromosome 15q11-q13 deletions, the critical PWS and AS regions (arrows) and the essential imprinting elements (AS-SRO and PWS-SRO) are shown. Maternal and paternal chromosomes are indicated. Genes expressed from the maternal allele are shown as pink boxes. Paternally expressed alleles are indicated by blue boxes. The silent, non-expressed allele is shown as a black box. Non-imprinted genes (i. e. expressed from both alleles) are in green. Imprinting of UBE3A, the sense transcript and the antisense transcript are tissue-specific. In the brain, the UBE3A and the sense transcript are expressed from the maternal allele while the antisense transcript is transcribed in the opposite orientation from the paternal allele. Other tissues do not express the antisense transcript but express UBE3A biallelically. The size of the antisense transcript has yet to be determined (light blue). PAR2 is contained within the UBE3A gene (10, 41). The expressed HERC2 pseudogenes are indicated by circles. | ||
Unequal crossover events between repeat sequences at 15q11
and 15q13 likely generate the large deletions observed in PWS
and AS (14, 15). The HERC2/Herc2 gene is unlikely to be imprinted
given that
1- it is expressed in somatic cell hybrids with a single maternal
or a single paternal chromosome 15, [n.
b. E' possibile ottenere degli ibridi che contengono come contributo
umano oltre ad altri cromosomi un solo dei due crom. 15. Se il
gene e' espresso dalle cellule in coltura si puo' dimostrare la
funzionalita' di entrambi gli alleli, Naturalmente tramite polimorfismi
si deve poter distinguere fra gli omologhi].
2- it is located in a non-imprinted region in humans and mice
and
3- mutations in murine Herc2 are inherited as a recessive trait
(13, 16).
In addition to large chromosomal deletions, smaller microdeletions
situated upstream of the SNRPN gene have been identified (17-21).
These localized deletions appear to disrupt the epigenetic program
that regulates imprinted gene expression across 15q11-q13, defining
a putative cis-acting imprinting control center (IC) (20). This
means that while chromosome 15 exhibits a normal biparental mode
of inheritance, AS patients have two chromosomes with a paternal
identity (hypomethylation and biallelic expression of paternally
expressed genes) and PWS patients have two chromosomes with a
maternal identity (hypermethylated and silent paternal genes).
Molecular characterization of the IC has established that this
region covers ~100 kb of genomic sequence and consists of a bipartite
structure (17, 19, 21). Deletion of the proximal portion of the
IC (25-30 kb upstream of the SNRPN promoter) results in AS (Fig.
1). Recently, the AS imprinting control element has been narrowed
to a region of 1. 15 kb [AS shortest region
of overlap (AS-SRO) cfr figura 4 dell'articolo Genetics of Agelman
syndrome]. (22). This element is hypothesized to be involved
in the imprinting process that establishes the maternal epigenotype
of 15q11-q13 (23, 24). In PWS, it is the distal portion of the
IC that is deleted. The PWS imprinting control element spans the
SNRPN promoter and exon 1 and is estimated to be <4. 3 kb in
size, as determined by the shortest region of overlap (PWS-SRO)
of microdeletions in PWS individuals [cfr
figura 4 dell'articolo: Genetics of Agelman syndrome] (25).
This element is hypothesized to function in the germline to establish
the paternal identity of 15q11-q13 by switching the grandmaternal
imprint to a paternal imprint (20, 24, 26). These imprinting elements
act to regulate imprinted expression across a domain of 2-3 Mb.
Although still poorly understood, several models have been proposed
for the role that the IC may play in the imprinting process (4,
20, 22-28).
CANDIDATE GENES FOR PWS
The PWS critical region extends over nearly half of 15q11-q13
and contains multiple paternally expressed genes. (Fig. 1). In
addition, several paternally expressed SNRPN upstream exons have
been localized to this region and are spliced in various combinations
to produce the IC transcripts (5, 10, 20, 29). Murine homologs
of these genes/transcripts map to the syntenic region of mouse
central chromosome 7 (5, 31-33).
Since the PWS critical region is so large it is likely that more
than one paternally expressed gene is involved in the pathogenesis
of PWS. However, it is uncertain which of these genes is involved
as no intragenic mutation affecting expression of only one PWS
gene has been described and loss of expression of a single specific
candidate gene has not been correlated with PWS. Some PWS patients
with rare balanced translocations show loss of expression of a
subset of paternally expressed genes while others exhibit normal
imprinted expression of these same genes (34-37). Recent identification
of a novel protein contained within the 5'-portion of the SNRPN
gene may help to explain these data (31). In two of the four translocation
patients the novel protein-encoding exons, termed SNURF (SNRPN
upstream reading frame), are severed while the SNRPN exons remain
intact (35, 37).
[n. b.: gli autori che hanno descritto uno
di questi pazienti portatore di una t(15; 19)de novo, hanno trovato
che il punto di rottura sul cromosoma 15 che era quello paterno,
cadeva fra l'esone 0 e l'esone 1 al di fuori della regione codificante
di SNRPN. Il prodotto del gene e' stato ritrovato intatto utilizzando
RT-PCR con primer degli'esoni 2 e 8, probabilmente perche' espresso
grazie all'azione di un regolatore del cromosoma 19 attivo su
cellule in coltura su cui si era traslocato. Inoltre hanno trovato
utilizzando primer degli esoni -1 e 0 il trascritto corrispondente
agli esoni -1, 0 che mappano sul derivativo 15. Quello che manca
in questo paziente e' proprio SNURF che e' codificato a partire
dall'esone -1, fino ad 8. Per chiarire questo guardare la figura
3 dell'articolo intitolato Conversione dell'imprinting, in cui
sono descrtti i CDNA che costituiscono i trascritti alternativi
del gene SNRPN. La figura 2 spiega i dati sopra descritti ricavati
da 35].
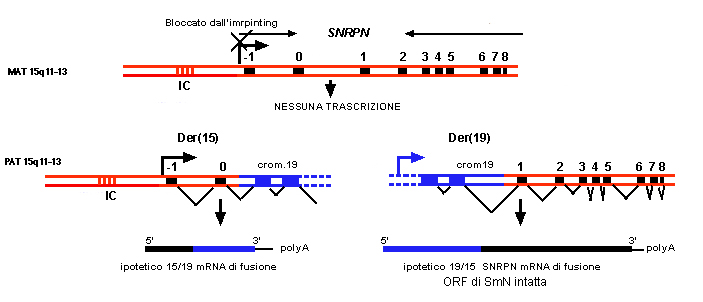
| Figura1. This locus has been renamed SNURF-SNRPN to depict the atypical, bicistronic nature of this gene (i. e. a single SNURF-SNRPN mRNA transcript from which the proteins SNURF and SmN [SmN e' uno splicing factor ed e' il prodotto della ORF dall'esone 1all'esone 8 ] are translated). SNURF-SNRPN is expressed from the paternal allele and, like SmN, SNURF protein is not detected in PWS patients. The SNURF exons are completely contained with the 4. 3 kb PWS-SRO, perhaps suggesting an important but not exclusive role for SNURF in the genesis of PWS: in the other translocation patients the breackpoint occurs downstream of the SNURF-SNRPN locus (34, 36). Currently, at best PWS can be characterized as a contiguous gene syndrome involving multiple paternally expressed genes. Complex mutant mice carrying targeted deletion of the various mouse homologs (see below) may identify which genes play a role in PWS. | ||
CANDIDATE GENES FOR AS
In contrast to PWS, ~20% of AS cases are predicted to have
intragenic mutations in the putative AS gene. The E6-AP ubiquitin-protein
ligase (UBE3A) gene has been strongly implicated as the AS gene
since genomic mutations and an inversion breakpoint have been
identified in UBE3A in AS patients (38-40). In addition, the entire
120 kb genomic UBE3A sequence is contained within the 250 kb AS
critical region (Fig. 1) (41). UBE3A/Ube3a exhibits tissue-specific
imprinting with preferential maternal expression in sub-regions
of the brain in humans and mice (42-45).
A fair number of AS patients have now been examined for the presence
of mutations in UBE3A. Only 30% of AS patients in this class had
loss-of-function mutations in UBE3A (46, 47). The remaining 70%
of patients had no identifiable defect in UBE3A. While this may
be explained by misdiagnosis of AS, it is also possible that additional
genes or silencing elements in the AS critical region are involved
in the pathogenesis of AS. Recently, additional transcripts have
been detected in this region, including a 3. 5 kb sense transcript
whose promoter is embedded in the 3'-UTR of the UBE3A gene (48).
This transcript is preferentially expressed from the maternal
allele in brain. Mutations in this candidate transcript/gene could
account for the remaining patients not possessing mutations in
UBE3A.
In addition to the sense transcript, an antisense transcript has also been identified (48). This transcript begins ~6. 5 kb from the UBE3A stop codon, includes sequences corresponding to the sense transcript and is coincident with the 3'-half of UBE3A. The size of this transcript has yet to be determined. In brain, the antisense transcript is expressed predominately from the paternal allele. A competition model has been proposed where transcription of the antisense gene would exclude paternal allele-specific UBE3A expression (4, 48).
EPIGENETIC MODIFICATION OF 15q11-q13
It is widely believed that the mechanism governing the imprinting of the PWS/AS domain is likely to involve parent-of-origin-specific epigenetic modification of the DNA. Studies have focused on epigenetic modifications such as allele-specific DNA methylation, replication timing and chromatin structure. SNRPN methylation patterns, which have been studied in the most detail, are likely to be an important part of the mechanism that controls imprinting at this locus. Both the human and mouse genes are hypermethylated on the inactive maternal allele at the promoter and first exon (corresponding to the PWS-SRO) and in the 3'-portion of the gene on the active paternal allele (18, 32, 52, 56-59). There is also evidence to suggest that these methylation patterns are established in the gametes, thereby representing candidate sequences for conferring the allelic imprinting mark (57, 58).
The well-established association between regulatory elements and nuclease hypersensitivity has led investigators to use chromatin analyses to search for regulatory elements. [ n. b. La sensibilita' alle nucleasi e' legata probabilmente al fatto che nei geni trascritti la cromatina e' meno condensanta per permettere l'azione dei fattori necessari alla trascrizione, rendendo il DNA piu' facilmente accessibile all'azione degli enzimi]. Consistent with the methylation patterns observed throughout the SNRPN locus, the promoter and exon 1 (PWS-SRO) are hypersensitive to nucleases on the paternal allele. While the paternal allele-specific hypersensitivity could merely reflect the transcriptionally active state of the SNRPN gene, it is also possible that this part of the IC is controlling the paternal-specific epigenotype. Interestingly, the AS-SRO is hypersensitive to nucleases on the maternal allele (60). Thus, the nuclease hypersensitivity of the PWS-SRO and AS-SRO in the IC supports the proposal that these regions serve to mediate the switching between paternal and maternal epigenotypes.
SNRPN and the PWS/AS region display other properties that are
characteristic of imprinted genes.
1- Many genes in the region harbor repetitive elements. For example,
the first intron of the mouse and human SNRPN genes contains structurally
conserved G-rich repeats (32, 59). As proposed for other imprinted
genes, the repeats may be involved in establishing the imprinting
or DNA methylation patterns of this gene (61).
2- The human chromosome 15 homologs replicate asynchronously and
exhibit preferential association during late S phase of the cell
cycle (62, 63).
MOUSE MODELS OF PWS AND AS
The first candidate mouse models for PWS and AS were described
by Cattanach et al. (65, 66). They used intercrosses between mice
harboring translocations to derive progeny with uniparental disomy
of the PWS/AS homologous region in mice. While these mice display
phenotypic characteristics indicative of the two syndromes, the
large region of uniparental disomy makes it difficult to assign
the phenotype to individual genes.
Yang et al. have generated a mouse model for PWS and IC mutations
by using homologous recombination in embryonic stem (ES) cells
to engineer a deletion of the Snprn gene and the region that corresponds
to the distal portion of the IC (including the PWS-SRO) (67).
Chimeric males with a mutation in their maternally derived allele
were not capable of reversing the maternal epigenotype of the
mutant allele in their germline and, as such, the progeny inheriting
this allele from the chimeric male failed to express the genes
normally transcribed exclusively from the paternal allele (i.
e. Snrpn, Zfp127, Ndn and Ipw). These mice displayed some of the
phenotypes characteristic of PWS. Thus, in addition to generating
a mouse model of PWS, this mutation mimics human IC mutations,
indicating that the position and hypothesized role of the IC are
conserved between mice and humans.
The mouse models and data from PWS patients provide compelling
evidence that perturbations in multiple paternally expressed genes
are involved in the pathogenesis of PWS. In agreement with this
proposal, mice that harbor an intragenic deletion of the Snrpn
gene are phenotypically normal (67). Thus, perturbations in Snrpn
gene expression alone are not sufficient to cause PWS symptoms
in the mouse. To prove that more than one gene is involved, mice
with mutations in multiple paternally expressed genes will have
to be derived.
ES cell technology has also been used to generate mutations in two candidate genes for AS, Ube3a and the b3 subunit of the GABAA receptor (Gabrb3). Mice with maternal deficiency of the imprinted Ube3a gene display a phenotype that mimics AS, including motor dysfunction, inducible seizures and a context-dependent learning deficit (45). Although absence of an imprinted gene that is expressed exclusively from the maternal allele (i. e. UBE3A) is the most likely AS candidate gene, the non-imprinted b3 subunit of the GABAA receptor (GABRB3) gene is located in the large deletion region of the majority of AS patients and may also contribute to the phenotype. Mice lacking the Gabrb3 gene exhibit seizures, learning and memory deficits, poor motor skills and hyperactivity, features that are common to AS (68, 69). Additionally, heterozygous Gabrb3 mutant mice exhibit a partial phenotype, suggesting that haploinsufficiency of the GABRB3 gene could be a contributing factor in AS deletion patients.
During the last year, many research contributions have advanced our understanding of the pathogenesis of PWS and AS. Identification of the HERC2 gene and pseudogenes provided a molecular explanation for 15q11-q13 being a hotspot for recombination and thereby generating one of the most common interstitial deletions in humans. The lack of pseudogenes also explains why the same chromosomal instability is not observed in the mouse.
Many questions arose from the data. A cursory glance at the
genetic maps reveals a paucity of genes/transcripts in mouse when
compared with humans. 1- Will the novel sense and antisense Ube3a
transcripts be found in mouse? More to the point, if the IC contains
information that confers the primary imprinting mark, will sequence
identity between humans and mice be revealed at the genomic level?
2- While sequence conservation is certainly an attractive idea,
no conserved cis-acting elements have yet been identified in other
imprinted genes. The generation of murine mutations that mimic
imprinting mutations in humans suggests functional conservation
of the IC. Will the upstream Snrpn exons that compose the IC transcripts
be conserved in the mouse?
3- Alternatively, does anonymous transcription play a role in
determining parental identity? Nuclease hypersensitivity studies
may help to determine whether a similar underlying chromatin structure
exists in the mouse. With the advent of murine PWS and AS models,
these questions may be addressed, providing greater understanding
of the complex molecular mechanism that governs PWS and AS imprinted
gene expression.
REFERENCES
1. Barlow, D. P. (1997) Competition-a common motif for the
imprinting mechanism. EMBO J., 16, 6899-6905.
2. Bartolomei, M. S. and Tilghman, S. M. (1997) Genomic imprinting
in mammals. Annu. Rev. Genet., 31, 493-525.
3. Constância, M., Pickard, B., Kelsey, G. and Reik, W.
(1998) Imprinting mechanisms. Genome Res., 8, 881-900.
4. Brannan, C. I. and Bartolomei, M. S. (1999) Mechanisms of genomic
imprinting. Curr. Opin. Genet. Dev., 9, 164-170.
5. Nicholls, R. D., Saitoh, S. and Horsthemke, B. (1998) Imprinting
in Prader-Willi and Angelman syndromes. Trends Genet., 14, 194-200.
6. Jiang, Y. -H., Tsai, T. -F., Bressler, J. and Beaudet, A. L.
(1998) Imprinting in Angelman and Prader-Willi syndromes. Curr.
Opin. Genet. Dev., 8, 334-342.
7. Clayton-Smith, J. and Pembrey, M. E. (1992) Angelman syndrome.
J. Med. Genet., 29, 412-415.
8. Cassidy, S. B. (1997) Prader-Willi syndrome. J. Med. Genet.,
34, 917-923.
9. Christian, S. L., Robinson, W. P., Huang, B., Mutirangura,
A., Line, M. R., Nakao, M., Surti, U., Chakravarti, A. and Ledbetter,
D. H. (1995) Molecular characterization of two proximal deletion
breakpoint regions in both Prader-Willi and Angelman syndrome
patients. Am. J. Hum. Genet., 57, 40-48.
10. Christian, S. L., Bhatt, N. K., Martin, S. A., Sutcliffe,
J. S., Kubota, T., Huang, B., Mutirangura, A., Chinault, A. C.,
Beaudet, A. L. and Ledbetter, D. H. (1998) Integrated YAC contig
map of the Prader-Willi/Angelman region on chromosome 15q11-13
with average STS spacing of 35 kb. Genome Res., 8, 146-157.
11. Kuwano, A., Mutirangura, A., Dittrich, B., Buiting, K., Horsthemke,
B., Saitoh, S., Niikawa, N., Ledbetter, S. A., Greenberg, F.,
Chinault, A. C. and Ledbetter, D. H. (1992) Molecular dissection
of the Prader-Willi/Angelman syndrome region (15q11-13) by YAC
cloning and FISH analysis. Hum. Mol. Genet., 1, 417-425.
12. Buiting, K., Groß, S., Ji, Y., Senger, G., Nicholls,
R. D. and Horsthemke, B. (1998) Expressed copies of the MN7 (D15F37)
gene family map close to the common deletion breakpoints in the
Prader-Willi/Angelman syndromes. Cytogenet. Cell Genet., 81, 247-253.
13. Ji, Y., Walkowica, M. J., Buiting, K., Johnson, D., Tarvin,
R. E., Rinchik, E. M., Horsthemke, B., Stubbs, L. and Nicholls,
R. D. (1999) The ancestral gene for transcribed, low-copy repeats
in the Prader-Willi/Angelman region encodes a large protein implicated
in protein trafficking, which is deficient in mice with neuromuscular
and spermiogenic abnormalities. Hum. Mol. Genet., 8, 533-542.
14. Carrozzo, R., Rossi, E., Christian, S. L., Kittikamron, K.,
Livieri, C., Corrias, A., Pucci, L., Fois, A., Simi, P., Bosio,
L., Beccaria, L., Zuffardi, O. and Ledbetter, D. H. (1997) Inter-
and intrachromosomal rearrangements are both involved in the origin
of 15q11-q13 deletions in Prader-Willi syndrome. Am. J. Hum. Genet.,
61, 228-231.
15. Robinson, W. P., Dutly, F., Nicholls, R. D., Bernasconi, F.,
Penaherrera, M., Michaelis, R. C., Abeliovich, D. and Schinzel,
A. A. (1998) The mechanisms involved in the formation of deletions
and duplications of 15q11-q13. J. Med. Genet., 35, 130-136.
16. Lehman, A. L., Nakatsu, Y., Ching, A., Bronson, R. T., Oakey,
R. J., Keiper-Hrynko, N., Finger, J. N., Durham-Pierre, D., Horton,
D. B., Newton, J. M., Lyon, M. F. and Brilliant, M. H. (1998)
A very large protein with diverse functional motifs is deficient
in rjs (runty, jerky, sterile) mice. Proc. Natl Acad. Sci. USA,
95, 9436-9441.
17. Reis, A., Dittrich, B., Greger, V., Buiting, K., Lalande,
M., Gillessen-Kaesbach, G., Anvret, M. and Horsthemke, B. (1994)
Imprinting mutations suggested by abnormal DNA methylation patterns
in familial Angelman and Prader-Willi syndromes. Am. J. Hum. Genet.,
54, 741-747.
18. Sutcliffe, J. S., Nakao, M., Christian, S., Orstavik, K. H.,
Tommerup, N., Ledbetter, D. H. and Beaudet, A. L. (1994) Deletions
of a differentially methylated CpG island at the SNRPN gene define
a putative imprinting control region. Nature Genet., 8, 52-58.
19. Buiting, K., Saitoh, S., Gross, S., Dittrich, B., Schwartz,
S., Nicholls, R. D. and Horsthemke, B. (1995) Inherited microdeletions
in the Angelman and Prader-Willi syndromes define an imprinting
centre on human chromosome 15. Nature Genet., 9, 395-400.
20. Dittrich, B., Buiting, K., Korn, B., Rickard, S., Buxton,
J., Saitoh, S., Nicholls, R. D., Poustka, A., Winterpacht, A.,
Zabel, B. and Horsthemke, B. (1996) Imprint switching on human
chromosome 15 may involve alternative transcripts of the SNRPN
gene. Nature Genet., 14, 163-170. 21. Saitoh, S., Buiting, K.,
Rogan, P. K., Buxton, J. L., Driscoll, D. J., Arnemann, J., Konig,
R., Malcolm, S., Horsthemke, B. and Nicholls, R. D. (1996) Minimal
definition of the imprinting center and fixation of a chromosome
15q11-q13 epigenotype by imprinting mutations. Proc. Natl Acad.
Sci. USA, 93, 7811-7815.
22. Ohta, T., Buiting, K., Kokkonen, H., McCandless, S., Heeger,
S., Leisti, H., Driscoll, D. J., Cassidy, S. B., Horsthemke, B.
and Nicholls, R. D. (1999) Molecular mechanism of Angelman syndrome
in two large families involves an imprinting mutation. Am. J.
Hum. Genet., 64, 385-396.
23. Bürger, J., Buiting, K., Dittrich, B., Groß, S.,
Lich, C., Sperling, K., Horsthemke, B. and Reis, A. (1997) Different
mechanisms and recurrence risks of imprinting defects in Angelman
syndrome. Am. J. Hum. Genet., 61, 88-93.
24. Buiting, K., Dittrich, B., Groß, S., Lich, C., Farber,
C., Buchholz, T., Smith, E., Reis, A., Burger, J., Nothen, M.
M., Barth-Witte, U., Janssen, B., Abeliovich, D., Lerer, I., van
den Ouweland, A., Halley, D. J. J., Schrander-Stumpel, C., Smeets,
H., Meinecke, P., Malcolm, S., Gardner, S., Lalande, M., Nicholls,
R. D., Friend, K., Schulze, A., Matthijs, G., Kokkonen, H., Hilbert,
P., Maldergem, L. V., Glover, G., Carbonell, P., Willems, P.,
Gillessen-Kaesbach, G. and Horsthemke, B. (1998) Sporadic imprinting
defects in Prader-Willi syndrome and Angelman syndrome: implications
for imprint-switch models, genetic counseling, and prenatal diagnosis.
Am. J. Hum. Genet., 63, 170-180.
25. Ohta, T., Gray, T. A., Rogan, P. K., Buiting, K., Gabriel,
J. M., Saitoh, S., Muralidhar, B., Bilienska, B., Krajewska-Walasek,
M., Driscoll, D. J., Horsthemke, B., Butler, M. G. and Nicholls,
R. D. (1999) Imprinting-mutation mechanism in Prader-Willi syndrome.
Am. J. Hum. Genet., 64, 397-413.
26. Ferguson-Smith, A. C. (1996) Imprinting moves to the center.
Nature Genet., 14, 119-121.
27. Schumacher, A., Buiting, K., Zeshnigk, M., Doerfler, W. and
Horsthemke, B. (1998) Methylation analysis of the PWS/AS region
does not support an enhancer-competition model. Nature Genet.,
19, 324-325.
28. Tilghman, S. M., Caspary, T. and Ingram, R. S. (1998) Competitive
edge at the imprinted Prader-Willi/Angelman region? Nature Genet.,
18, 206-208.
29. Ning, Y., Roschke, A., Christian, S., Lesser, J., Sutcliffe,
J. S. and Ledbetter, D. H. (1996) Identification of a novel paternally
expressed transcript adjacent to snRPN in the Prader-Willi syndrome
critical region. Genome Res., 6, 742-746.
30. Jong, M. T., Gray, T. A., Ji, Y., Glenn, C. C., Saitoh, S.,
Driscoll, D. J. and Nicholls, R. D. (1999) A novel imprinted gene,
encoding a RING zinc-finger protein, and overlapping antisense
transcript in the Prader-Willi syndrome critical region. Hum.
Mol. Genet., 8, 783-793.
31. Gray, T. A., Saitoh, S. and Nicholls, R. D. (1999) An imprinted,
mammalian bicistronic transcript encodes two independent proteins.
Proc. Natl Acad. Sci. USA, 96, 5616-5621.
32. Gabriel, J. M., Gray, T. A., Stubbs, L., Saitoh, S., Ohta,
T. and Nicholls, R. D. (1998) Structure and function correlations
at the imprinted mouse Snrpn locus. Mamm. Genome, 9, 788-793.
33. Jong, M. T., Carey, A. H., Caldwell, K. A., Lau, M. H., Handel,
M. A., Driscoll, D. J., Steward, C. L., Rinchik, E. M. and Nicholls,
R. D. (1999) Imprinting of a RING zinc-finger encoding gene in
the mouse chromosome region homologous to the Prader-Willi syndrome
genetic region. Hum. Mol. Genet., 8, 795-803.
34. Schulze, A., Hansen, C., Skakkebaek, N. E., Brondum-Nielsen,
K., Ledbetter, D. H. and Tommerup, N. (1996) Exclusion of SNRPN
as a major determinant of Prader-Willi syndrome by a translocation
breakpoint. Nature Genet., 12, 452-454.
35. Sun, Y., Nicholls, R. D., Butler, M. G., Saitoh, S., Hainline,
B. E. and Palmer, C. G. (1996) Breakage in the SNRPN locus in
a balanced 46, XY, t(15; 19) Prader-Willi syndrome patient. Hum.
Mol. Genet., 5, 517-524.
36. Conroy, J. M., Grebe, T. A., Becker, L. A., Tsuchiya, K.,
Nicholls, R. D., Buiting, K., Horsthemke, B., Cassidy, S. B. and
Schwartz, S. (1997) Balanced translocation 46, XY, t(2; 15)(q37.
2; q11. 2) associated with atypical Prader-Willi syndrome. Am.
J. Hum. Genet., 61, 388-394.
37. Kuslich, C. D., Kobori, J. A., Mohapatra, G., Gregorio-King,
C. and Donlon, T. A. (1999) Prader-Willi syndrome is caused by
disruption of the SNRPN gene. Am. J. Hum. Genet., 64, 70-76.
38. Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J.,
Jiang, Y. H., Benton, C. S., Rommens, J. M. and Beaudet, A. L.
(1997) De novo truncating mutations in E6-AP ubiquitin-protein
ligase gene (UBE3A) in Angelman syndrome. Nature Genet., 15, 74-77.
39. Kishino, T., Lalande, M. and Wagstaff, J. (1997) UBE3A/E6-AP
mutations cause Angelman syndrome. Nature Genet., 15, 70-73.
40. Greger, V., Knoll, J. H., Wagstaff, J., Woolf, E., Lieske,
P., Glatt, H., Benn, P. A., Rosengren, S. S. and Lalande, M. (1997)
Angelman syndrome associated with an inversion of chromosome 15q11.
2q24. 3. Am. J. Hum. Genet., 60, 574-580.
41. Sutcliffe, J. S., Jiang, Y., Galjaard, R., Matsuura, T., Fang,
P., Kubota, T., Christian, S. L., Bressler, J., Cattanach, B.,
Ledbetter, D. H. and Beaudet, A. L. (1997) The E6-AP ubiquitin-protein
ligase (UBE3A) gene is localized within a narrowed Angelman syndrome
critical region. Genome Res., 7, 368-377.
42. Rougeulle, C., Glatt, H. and Lalande, M. (1997) The Angelman
syndrome candidate gene, UBE3A/E6-AP, is imprinted in the brain.
Nature Genet., 17, 14-15.
43. Vu, T. H. and Hoffman, A. R. (1997) Imprinting of the Angelman
syndrome gene, UBE3A, is restricted to the brain. Nature Genet.,
17, 12-13.
44. Albrecht, J., Sutcliffe, J. S., Cattanach, B., Beechey, C.
V., Armstrong, D., Eichele, G. and Beaudet, A. L. (1997) Imprinted
expression of the murine Angelman syndrome gene, Ube3a, in hippocampal
and Purkinje neurons. Nature Genet., 17, 75-78.
45. Jiang, Y., Armstrong, D., Albrecht, U., Atkins, C., Noebels,
J. L., Eichele, G., Sweatt, J. D. and Beaudet, A. L. (1998) Mutation
of the Angelman ubiquitin ligase in mice causes increased cytoplasmic
p53 and deficits of contextual learning and long-term potentiation.
Neuron, 21, 799-811.
46. Malzac, P., Webber, H., Moncla, A., Graham, J. M. Jr, Kukolich,
M., Williams, C., Pagon, R. A., Ramsdell, L. A., Kishino, T. and
Wagstaff, J. (1998) Mutation analysis of UBE3A in Angelman syndrome
patients. Am. J. Hum. Genet., 62, 1353-1360.
47. Fang, P., Lev-Lehman, E., Tsai, T. -F., Matsuura, T., Benton,
C. S., Sutcliffe, J. S., Christian, S. L., Kubota, T., Halley,
D. J., Meijers-Heijboer, H., Langlois, S., Graham, J. M., Jr,
Beuten, J., Willems, P. J., Ledbetter, D. H. and Beaudet, A. L.
(1999) The spectrum of mutations in UBE3A in Angelman syndrome.
Hum. Mol. Genet., 8, 129-135.
48. Rougeulle, C., Cardoso, C., Fontés, M., Colleaux, L.
and Lalande, M. (1998) An imprinted antisense RNA overlaps UBE3A
and a second maternally expressed transcript. Nature Genet., 19,
15-16.
49. Driscoll, D. J., Waters, M. F., Williams, C. A., Zori, R.
T., Glenn, C. C., Avidano, K. M. and Nicholls, R. D. (1992) DNA
methylation imprint, determined by the sex of the parent, distinguishes
Angelman and Prader-Willi syndromes. Genomics, 13, 917-924.
50. Dittrich, B., Buiting, K., Groß, S. and Horsthemke,
B. (1993) Characterization of a methylation imprint in the Prader-Willi
syndrome chromosome region. Hum. Mol. Genet., 2, 1995-1999.
51. Glenn, C. C., Nicholls, R. D., Robinson, W., Saitoh, S., Niikawa,
N., Schinzel, A., Horsthemke, B. and Driscoll, D. J. (1993) Modification
of 15q11-q13 DNA methylation imprints in unique Angelman and Prader-Willi
patients. Hum. Mol. Genet., 2, 1377-1382.
52. Zeschnigk, M., Schmitz, B., Dittrich, B., Buiting, K., Horsthemke,
B. and Doerfler, W. (1997) Imprinted segments in the human genome:
different DNA methylation patterns in the Prader-Willi/Angelman
syndrome region as determined by the genomic sequencing method.
Hum. Mol. Genet., 6, 387-395.
53. LaSalle, J. M., Ritchie, R. J., Glatt, H. and Lalande, M.
(1998) Clonal heterogeneity at allelic methylation sites diagnostic
for Prader-Willi and Angelman syndromes. Proc. Natl Acad. Sci.
USA, 95, 1675-1680. 54. Jay, P., Rougeulle, C., Massacrier, A.,
Moncla, A., Mattei, M. -G., Malzac, P., Roeckel, N., Taviaux,
S., Lefranc, J. -L. B., Cau, P., Berta, P., Lalande, M. and Muscatelli,
F. (1997) The human necdin gene, NDN, is maternally imprinted
and located in the Prader-Willi syndrome chromosomal region. Nature
Genet., 17, 357-361.
55. Watrin, F., Roaeckel, N., Lacroix, L., Mignon, C., Mattei,
M. G., Disteche, C. and Muscatelli, F. (1997) The mouse Necdin
gene is expressed from the paternal allele only and lies in the
7C region of mouse chromosome 7, a region of conserved synteny
to the human Prader-Willi syndrome region. Eur. J. Hum. Genet.,
5, 324-332.
56. Glenn, C. C., Porter, K. A., Jong, M. T. C., Nicholls, R.
D. and Driscoll, D. J. (1993) Functional imprinting and epigenetic
modification of the human SNRPN gene. Hum. Mol. Genet., 2, 2001-2005.
57. Glenn, C. C., Saitoh, S., Jong, M. T. C., Filbrandt, M. M.,
Surti, U., Driscoll, D. J. and Nicholls, R. D. (1996) Gene structure,
DNA methylation, and imprinted expression of the human SNRPN gene.
Am. J. Hum. Genet., 58, 335-346.
58. Shemer, R., Birger, Y., Riggs, A. D. and Razin, A. (1997)
Structure of the imprinted mouse Snrpn gene and establishment
of its parental-specific methylation pattern. Proc. Natl Acad.
Sci. USA, 94, 10267-10272.
59. Blaydes, S. M., Elmore, M., Yang, T. and Brannan, C. I. (1999)
Analysis of murine Snrpn and human SNRPN imprinting in transgenic
mice. Mamm. Genome, 10, 549-555.
60. Schweizer, J., Zynger, D. and Francke, U. (1999) In vivo nuclease
hypersensitivity studies reveal multiple sites of parental origin-dependent
differential chromatin conformation in the 150 kb SNRPN transcription
unit. Hum. Mol. Genet., 8, 555-566.
61. Neumann, B., Kubicka, P. and Barlow, D. P. (1995) Characteristics
of imprinted genes. Nature Genet., 9, 12-13.
62. Kitsberg, D., Selig, S., Brandeis, M., Simon, I., Keshet,
I., Driscoll, D. J., Nicholls, R. D. and Cedar, H. (1993) Allele-specific
replication timing of imprinted gene regions. Nature, 364, 459-463.
63. LaSalle, J. M. and Lalande, M. (1996) Homologous association
of oppositely imprinted chromosomal domains. Science, 272, 725-728.
64. Lyko, F., Buiting, K., Horsthemke, B. and Paro, R. (1998)
Identification of a silencing element in the human 15q11-q13 imprinting
center by using transgenic Drosophila. Proc. Natl Acad. Sci. USA,
95, 1698-1702.
65. Cattanach, B. M., Barr, J. A., Evans, E. P., Burtenshaw, M.,
Beechey, C. V., Leff, S. E., Brannan, C. I., Copeland, N. G.,
Jenkins, N. A. and Jones, J. (1993) A candidate mouse model for
Prader-Willi syndrome which shows an absence of Snrpn expression.
Nature Genet., 2, 270-274.
66. Cattanach, B. M., Barr, J. A., Beechey, C. V., Martin, J.,
Noebels, J. and Jones, J. (1997) A candidate model for Angelman
syndrome in the mouse. Mamm. Genome, 8, 472-478.
67. Yang, T., Adamson, T. E., Resnick, J. L., Leff, S., Wevrick,
R., Francke, U., Jenkins, N. A., Copeland, N. G. and Brannan,
C. I. (1998) A mouse model for Prader-Willi syndrome imprinting-centre
mutations. Nature Genet., 19, 25-31.
68. Homanics, G. E., Delorey, T. M., Firestone, L. L., Quinlan,
J. J., Handforth, A., Harrison, N. L., Krasowski, M. D., Rick,
C. E., Korpi, E. R., Makela, R., Brilliant, M. H., Hagiwara, N.,
Ferguson, C., Snyder, K. and Olsen, R. W. (1997) Mice devoid of
[gamma]-aminobutyrate type A receptor [beta]3 subunit have epilepsy,
cleft palate and hypersensitive behavior. Proc. Natl Acad. Sci.
USA, 94, 4143-4148.
69. DeLorey, T. M., Handforth, A., Anagnostaras, S. G., Homanics,
G. E., Minassian, B. A., Asatourian, A., Fanselow, M. S., Delagado-Escueta,
A., Ellison, G. D. and Olsen, R. W. (1998) Mice lacking the [beta]3
subunit of the GABAA receptor have the epilepsy phenotype and
many of the behavioral characteristics of Angelman syndrome. J.
Neurosci., 18, 8505-8514.
70. Nicholls, R. D., Gottlieb, W., Russell, L. B., Davda, M.,
Horsthemke, B. and Rinchik, E. M. (1993) Evaluation of potential
models for imprinted and nonimprinted components of human chromosome
15q11-q13 syndromes by fine-structure homology mapping in the
mouse. Proc. Natl Acad. Sci. USA, 90, 2050-2054.
71. Johnson, D. K., Stubbs, L. J., Culiat, C. T., Montgomery,
C. S., Russell, L. B. and Rinchik, E. M. (1995) Molecular analysis
of 36 mutations at the mouse pink-eye dilution (p)locus. Genetics,
141, 1563-1571.